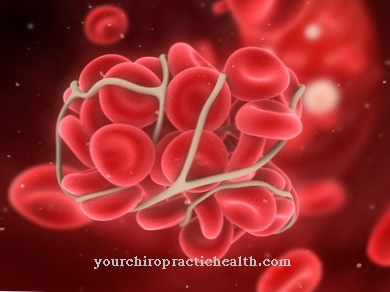
Po urazie krwotocznym Agregacja płytek krwi powrót do zdrowia jako ważny krok w leczeniu ran. Zapewnia, że płytki krwi gromadzą się w ciągu kilku minut, sklejają ze sobą uszkodzony obszar iw ten sposób odpływają krew.
Co to jest agregacja płytek krwi?

Agregacja płytek krwi to nazwa nadana istotnej części procesu podczas krzepnięcia krwi. W pierwszej fazie krzepnięcia krwi (hemostaza pierwotna) płytki krwi (gr. Trombos, grudki) zapewniają pierwotne zamknięcie rany poprzez zlepianie się i gromadzenie (łac. Agregare, kumulacja).
Procesowi towarzyszy zwężenie zajętych naczyń krwionośnych i okolic. Płytki krwi (trombocyty) zmieniają swój wygląd i właściwości na powierzchni komórki, gdy się zlepiają. Zmiana kształtu płytek krwi odsłania receptory na powierzchni, które są teraz aktywne. Aktywowane płytki krwi mogą przez nie przylegać do ściany naczynia.
Ponadto zachodzą inne procesy wspomagające hemostazę. Wszelkie czynniki uwalniane na uszkodzonej ścianie naczynia kierują płytki krwi do tego punktu. Ponadto uwalniane są substancje, które zapobiegają stanom zapalnym i inicjują kolejne kroki w krzepnięciu krwi. Zapewniają trwałe zamknięcie rany i ostateczne wygojenie.
Funkcja i zadanie
Agregacja płytek krwi zapobiega znacznej utracie krwi po urazie. Ten proces jest częścią układu krzepnięcia krwi. System ten działa jako złożona i precyzyjnie dostrojona gra różnych komórek (płytek krwi), czynników krzepnięcia i kilku substancji przekaźnikowych. Działa jak reakcja łańcuchowa.
Czynniki krzepnięcia to głównie białka, które są aktywowane w określonych warunkach i z kolei inicjują lub przyspieszają reakcje w procesie krzepnięcia. W zastosowaniach medycznych czynnikom krzepnięcia nadano liczby rzymskie (od 1 do 13).
Płytki krwi inicjują reakcję koagulacji, jeśli powierzchnia jest uszkodzona. Proces, który za tym stoi, przebiega w trzech fazach. Adhezja (łac. Przyklejać), a także agregacja płytek krwi i tworzenie się czopa zamykającego ranę. Ściany komórkowe uszkodzonych naczyń lub tkanek uwalniają czynnik koagulacyjny, tak zwany czynnik von Willebranda. Jest to cząsteczka białka, która jest syntetyzowana przez komórki ściany wewnętrznej naczynia (komórki śródbłonka) i komórki prekursorowe płytek krwi. Jest przechowywany w płytkach krwi i uwalniany po aktywacji. Czynnik ten odpowiada za adhezję płytek krwi (przyleganie do ściany naczynia), tak aby cienko pokrywały ranę.
Jednocześnie w ten sposób inicjowana jest agregacja płytek krwi. Dzieje się tak, ponieważ po aktywacji płytek krwi aktywowane są również geny, które wprawiają w ruch syntezę receptora niezbędnego do agregacji. Za pomocą strukturalnego białka kolagenu, trombiny, ważnego enzymu krzepnięcia krwi, nukleotydu adenozynodifosforanu (ADP), hormonów, takich jak adrenalina i inne substancje endogenne, płytki krwi zmieniają swój kształt. W trakcie tego procesu uwalniane są dalsze składniki, a dotknięty obszar jest przygotowywany do kolejnych etapów krzepnięcia krwi.
Uaktywnia się kaskada różnych czynników. Podczas gdy agregacja płytek jest początkowo odwracalna, ostatecznie osiągany jest poziom, na którym tworzy się sieć płytek krwi z udziałem specjalnego białka (fibrynogenu, czynnik I) i nieodwracalnego skrzepliny (czop krzepnięcia krwi).
Choroby i dolegliwości
Zaburzenia agregacji płytek krwi mogą objawiać się nasileniem lub osłabieniem reakcji. Mogą wystąpić u pacjentów z dziedziczną predyspozycją lub po przyjęciu niektórych leków. Choroby wrodzone są rzadkie i wpływają na samą agregację płytek lub różne procesy towarzyszące temu procesowi.
Osoby dotknięte chorobą uwidaczniają się samoistnie występującym krwawieniem z błon śluzowych i nosa oraz skłonnością do krwiaków (siniaków). Kobiety cierpią na obfite krwawienia miesiączkowe i krwawienia porodowe.
Jedna z tych wad wrodzonych została nazwana na cześć szwajcarskiego pediatry E. Glanzmanna: choroba Glanzmanna-Naegeli (również trombastenia Glanzmanna). Jest dziedziczona jako cecha autosomalna recesywna. Dotknięty jest receptor w błonie płytek krwi, który nie jest udostępniany w wystarczających ilościach z powodu zmiany genetycznej (mutacji). Pacjenci z tą wadą są narażeni na duże ryzyko przyjmowania inhibitorów agregacji płytek krwi, takich jak Aspirin®.
W zespole Willebranda-Jürgensa czynnik ważny dla adhezji i agregacji płytek krwi nie występuje w wystarczających ilościach lub ma ograniczenia jakościowe. Oznacza to, że nie jest w pełni funkcjonalny, a adhezja płytek krwi jako etap przygotowawczy agregacji jest osłabiona.
Dwóch francuskich hematologów jest imiennikiem innej dziedzicznej, bardzo rzadkiej choroby płytek krwi: zespołu Bernarda-Souliera. Przede wszystkim wpływa to na adhezję płytek krwi. Zmniejsza się iw ten sposób zmniejsza również agregację płytek krwi.
Pacjenci z chorobą puli pamięci wykazują upośledzoną wydajność wydzielania po aktywacji płytek krwi. Wynika to z brakujących granulek. Są to złogi komórkowe (pęcherzyki), z których podczas aktywacji płytek krwi uwalniane są różne czynniki. Zespół szarych płytek krwi (zespół szarych płytek krwi) jest szczególną postacią.
Nabyte lub lecznicze zaburzenia agregacji płytek są również częściej rozpoznawane. Tak zwane wyczerpane płytki krwi, które nie są już zdolne do agregacji, mogą wystąpić u pacjentów dializowanych, przez płuco-serce, w ciężkich chorobach nerek lub po oparzeniach. Sytuacja w tych przypadkach jest podobna do choroby puli pamięci.
Zwiększoną agregację płytek stwierdza się w chorobie niedokrwiennej serca, po udarach, chorobach naczyń i ostrej zakrzepicy. W profilaktyce zakrzepicy często stosuje się leki hamujące czynność płytek krwi. Jednym z nich jest kwas acetylosalicylowy (np. W aspirynie). Istnieją również leki stosowane w chemioterapii, które zmniejszają agregację płytek krwi.








.jpg)


.jpg)