Homocysteina jest nieproteogennym alfa-aminokwasem zawierającym siarkę, który powstaje jako związek pośredni z metioniny przez uwolnienie grupy metylowej (-CH3).
Do dalszego przetwarzania homocysteiny niezbędna jest odpowiednia podaż witamin B12 i B6 oraz kwasu foliowego lub betainy jako dostawcy grup metylowych. Zwiększone stężenie homocysteiny w osoczu krwi wiąże się z uszkodzeniem ścian naczyń krwionośnych, otępieniem i depresją.
Co to jest homocysteina?
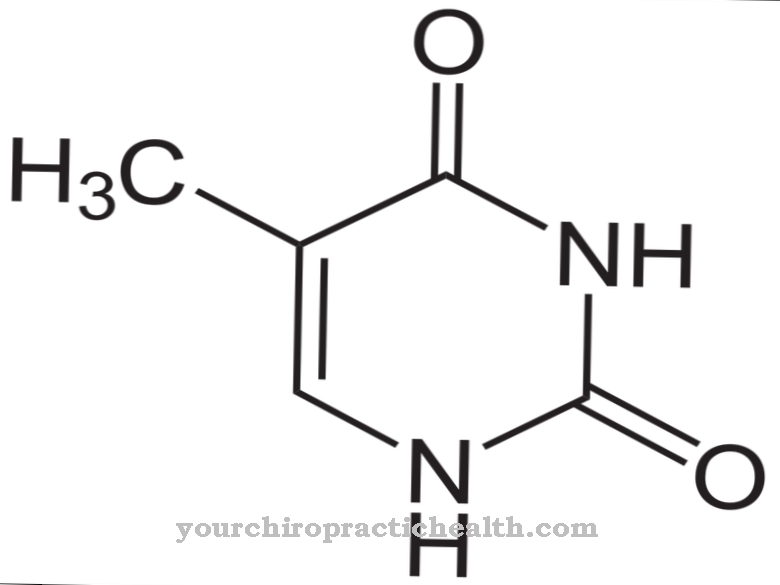
Homocysteina w swojej bioaktywnej postaci L jest aminokwasem nieproteogennym. Nie może być budulcem białka, ponieważ ma tendencję do tworzenia pierścienia heterocyklicznego, który nie pozwala na stabilne wiązanie peptydowe ze względu na dodatkową grupę CH2 w porównaniu z cysteiną.
Włączenie homocysteiny do białka spowodowałoby zatem szybki rozkład białka. Wzór chemiczny C4H9NO2S pokazuje, że aminokwas składa się wyłącznie z substancji, które są dostępne w dużych ilościach prawie wszędzie. Do ich budowy nie są wymagane pierwiastki śladowe, rzadkie minerały i metale. Homocysteina jest jonem obojnaczym, ponieważ ma dwie grupy funkcyjne, każda z ładunkiem dodatnim i ujemnym, które są ogólnie zrównoważone elektrycznie.
W temperaturze pokojowej homocysteina jest krystaliczną substancją stałą o temperaturze topnienia około 230 do 232 stopni Celsjusza. Organizm może rozłożyć podwyższony poziom homocysteiny we krwi przez dwie cząsteczki homocysteiny tworzące mostek dwusiarczkowy, tworząc homocystynę, która może być następnie wydalana przez nerki.
Funkcja, efekt i zadania
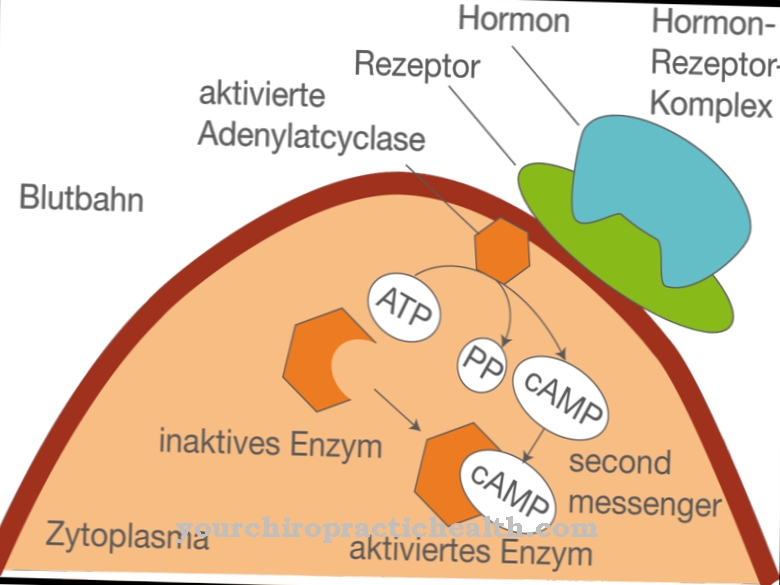
Najważniejszym zadaniem i funkcją L-homocysteiny jest wspomaganie syntezy białek i przekształcanie się w S-adenozylometioninę (SAM) we współpracy z niektórymi koenzymami. Z trzema grupami metylowymi (-CH3) SAM jest najważniejszym donorem grup metylowych w metabolizmie komórkowym.
SAM bierze udział w wielu reakcjach biosyntezy i detoksykacji. Grupy metylowe niektórych neuroprzekaźników, takich jak adrenalina, cholina i kreatyna, pochodzą z SAM. Po uwolnieniu grupy metylowej SAM przekształca się w S-adenozylometioninę (SAH), która jest ponownie przekształcana w adenozynę lub L-homocysteinę przez hydrolizę. Równie ważne jak wspomagająca funkcja homocysteiny dla pewnych procesów metabolicznych jest również to, że homocysteina, jako produkt pośredni tych reakcji biochemicznych i łańcuchów syntezy, nie występuje we krwi w nieprawidłowych stężeniach, ponieważ wywołuje wtedy szkodliwe skutki.
Nadmiar homocysteiny, który nie jest niezbędny do wspomagania opisanych powyżej reakcji metabolizmu metioniny, jest zatem normalnie dalej rozkładany z udziałem witaminy B6 (pirydoksyny) i wydalany przez nerki po utworzeniu homocystyny. Aby homocysteina mogła spełniać swoje zadania metaboliczne, ważne jest dostarczanie organizmowi wystarczającej ilości witamin B6, B12 i kwasu foliowego.
Edukacja, występowanie, właściwości i optymalne wartości
Homocysteina jest wytwarzana w organizmie jako krótkotrwały produkt pośredni w złożonym metabolizmie metioniny. Alternatywne oznaczenie kwasu (S) -2-amino-4-merkaptobutanowego wskazuje na budowę homocysteiny. Jest więc kwasem monokarboksylowym z charakterystyczną grupą karboksylową (-COOH) i jednocześnie prostym kwasem tłuszczowym. Homocysteina nie jest wchłaniana z pożywieniem, a jedynie tymczasowo wytwarzana w organizmie.
Chociaż bioaktywna L-cysteina odgrywa ważną rolę w syntezie białek i tworzeniu SAM, optymalne i jednocześnie tolerowane stężenie we krwi mieści się w wąskich granicach zaledwie 5 do 10 µmol / litr. Wyższe poziomy homocysteiny wskazują na pewne zaburzenia metaboliczne i prowadzą do klinicznego obrazu hiperhomocysteinemii. Optymalne stężenie aminokwasu prawdopodobnie zależy od odpowiedniej aktywności umysłowej i fizycznej i jest trudne do zdefiniowania. Bardziej sensowna wydaje się definicja tolerowanej górnej granicy poziomu homocysteiny, która powinna wynosić około 10 µmol / litr.
Choroby i zaburzenia
Jeśli stężenie homocysteiny przekracza dopuszczalną granicę, w równowadze metioninowej występują głównie nabyte lub uwarunkowane genetycznie zaburzenia metaboliczne.
Często brakuje tylko niezbędnych witamin B6 (pirydoksyna), B9 (kwas foliowy) i B12 (kobalamina), które są wymagane jako koenzymy lub katalizatory w łańcuchu konwersji biochemicznej. W sumie znanych jest około 230 - aczkolwiek rzadko występujących - mutacji genów, które prowadzą do zakłócenia metabolizmu metioniny. Patologiczny wzrost homocysteiny nazywa się homocystynurią. Najczęstsza mutacja genu powodująca chorobę znajduje się w locus genu 21q22.3. Mutacja jest autosomalna recesywna i powoduje powstanie wadliwego enzymu, który jest niezbędny do procesu rozkładu i konwersji homocysteiny.
Wcześniej znane mutacje to pominięcie (delecja) lub dodanie (insercja) zasad nukleinowych na odpowiednich niciach DNA. Niekorzystne warunki życia i nawyki mogą również powodować wzrost poziomu homocysteiny. Należą do nich nadmierne spożycie alkoholu, nadużywanie nikotyny, nadwaga i siedzący tryb życia. Nadmierny poziom homocysteiny może uszkodzić śródbłonek, wewnętrzną ścianę naczyń krwionośnych i B. Promuj miażdżycę. Żyły stają się nieelastyczne i powodują szereg wtórnych chorób, takich jak nadciśnienie. Niosą również ryzyko tworzenia się skrzeplin, które powodują chorobę wieńcową serca i udary.
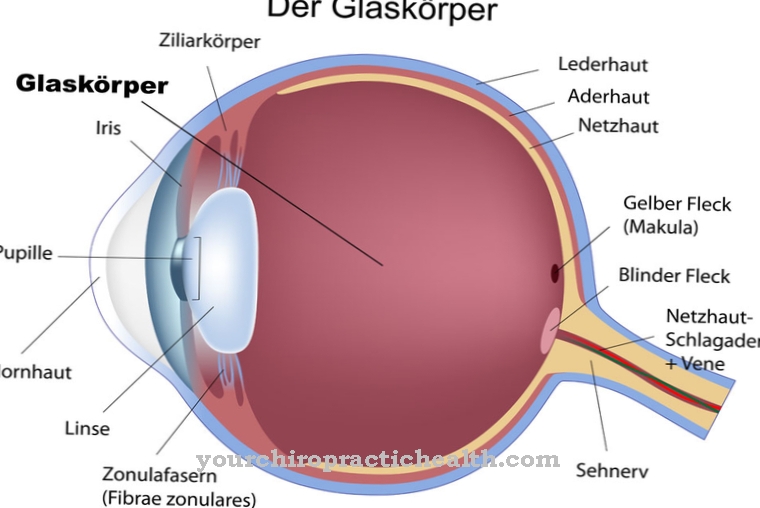
Choroby neurologiczne, takie jak depresja i demencja starcza, są również związane ze zwiększonym poziomem homocysteiny. U dzieci z genetyczną homocystynurią objawy choroby są bardzo różne. Spektrum objawów sięga od ledwo wykrywalnych cech choroby do pojawienia się prawie wszystkich możliwych objawów. Pierwsze objawy pojawiają się zwykle dopiero po ukończeniu drugiego roku życia. Co najwyżej spowolnienie rozwoju psychomotorycznego można zauważyć w pierwszych dwóch latach życia. W wielu przypadkach pierwszym objawem genetycznej homocystynurii jest wypadnięcie soczewki oka.

.jpg)



.jpg)




.jpg)