Tak jak Mitochondrium jest terminem używanym do opisania organelli komórkowych, które oprócz innych funkcji biorą udział przede wszystkim w dostarczaniu energii do komórki poprzez metabolizm adenozynotrifosforanu. Mitochondria posiadają własny materiał genetyczny w postaci mitochondrialnego DNA. W zależności od wymagań energetycznych typów komórek, w komórce może znajdować się od kilku do kilku tysięcy mitochondriów.
Co to jest mitochondrium?
Mitochondrium to organellum komórkowe, które znajduje się praktycznie we wszystkich ludzkich komórkach np. T. występuje duża liczba do kilku tysięcy. Wyjątkiem jest wierzchnia warstwa skóry, warstwa rogowa naskórka, która składa się z martwych komórek rogówki i nie zawiera mitochondronów.
Mitochondria są wyposażone we własny genom, mitochondrialne DNA (mtDNA), co potwierdza założenie, że mitochondria były pierwotnie niezależnymi organizmami, które weszły w endosymbiozę z komórkami wielokomórkowych żywych istot. Mitochondria nie są już w stanie samodzielnie żyć w swojej obecnej formie. Mitochondria charakteryzują się podwójną membraną, zewnętrzną, prawie gładką membraną i wewnętrzną, silnie pofałdowaną membraną, która zapewnia odpowiednio dużą powierzchnię dla biochemicznych procesów metabolicznych.
Między innymi mitochondria biorą udział w metabolizmie tzw. Łańcucha oddechowego i cyklu kwasu cytrynowego. W łańcuchu oddechowym, który przebiega w przestrzeni międzybłonowej między błoną zewnętrzną i wewnętrzną, glukoza jest metabolizowana w celu syntezy ATP i udostępniana komórce jako nośnik energii. W cyklu kwasu cytrynowego dochodzi do zjednoczenia procesów metabolicznych, które powodują rozpad węglowodanów, białek i tłuszczów.
Anatomia i budowa
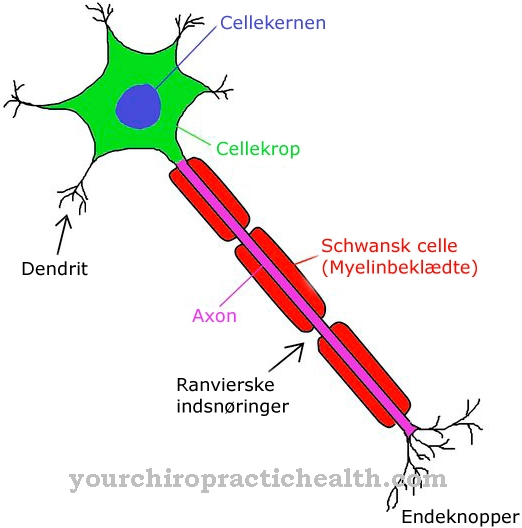
Dwie błony są charakterystyczne dla morfologii mitochondriów: błona zewnętrzna, która nadaje organelli kształt prawie fasolowy, oraz błona wewnętrzna, która jest silnie pofałdowana i dlatego ma dużą powierzchnię. Obie błony zbudowane są z dwuwarstw fosfolipidowych i białek. Jednak właściwości membrany zewnętrznej różnią się znacznie od membrany wewnętrznej.
Błona zewnętrzna zawiera kompleksy białkowe z kanałami, które umożliwiają selektywną wymianę substancji między mitochondrium a cytozolem komórki. Błona wewnętrzna mitochondriów typu sacculus zawiera kompleksy białek, które są niezbędne do „funkcjonowania” łańcucha oddechowego. Przestrzenie tworzone przez fałdy błony wewnętrznej w kierunku błony zewnętrznej są znane jako cristae i przyspieszają metabolizm łańcucha oddechowego.
Cristae pokryte są maleńkimi ciałkami o średnicy 8,5 nm, które są znane jako cząstki F1 lub cząstki syntazy ATP i odgrywają rolę w syntezie ATP. Innym rodzajem mitochondriów jest typ kanalików, który znajduje się w komórkach syntetyzujących hormony steroidowe. Liczne kanaliki służą do selektywnego transportu substancji.
Funkcja i zadania
Jedną z najważniejszych funkcji i zadań mitochondriów jest synteza trójfosforanu adenozyny (ATP) i uwalnianie ATP do macierzy komórkowej, wnętrza komórki poza mitochondriami. W złożonym łańcuchu reakcji energia uzyskana w kontrolowanych katalitycznie procesach utleniania jest krótko magazynowana w postaci ATP i udostępniana komórkom. Łańcuch oddechowy opiera się na produktach przemiany materii tzw. Cyklu kwasu cytrynowego - zwanego również cyklem Krebsa od odkrywcy Hansa A. Krebsa.
Metabolizm cyklu Krebsa zachodzi w macierzy mitochondriów, czyli w przestrzeni zamkniętej wewnętrzną błoną. Mitochondria biorą również udział w części cyklu mocznikowego, który zachodzi częściowo w macierzy mitochondrialnej, a częściowo w cytozolu komórki. Cykl mocznikowy służy do usuwania takich produktów rozkładu zawierających azot. B. zamienić pokarm zawierający białko na mocznik i wydalić go przez nerki. Mitochondria biorą również udział w procesie kontrolowanej śmierci komórek, czyli apoptozy.
Jest to rodzaj samozniszczenia komórki z uporządkowanym usuwaniem produktów degradacji. Apoptoza może np. B. być „nakazanym” przez układ odpornościowy w przypadku wykrycia poważnych defektów lub infekcji, aby zapobiec uszkodzeniom i niebezpiecznym sytuacjom dla całego organizmu. Mitochondria mają zdolność wychwytywania jonów wapnia i udostępniania ich komórce w razie potrzeby. Wspierają ważną funkcję homeostazy wapnia w komórce.
Inną ważną funkcją mitochondriów jest synteza skupisk żelaza i siarki, które są niezbędne dla wielu enzymów do katalitycznej kontroli łańcucha oddechowego. Synteza klastrów żelazo-siarka nie jest zbędna, dlatego jest niezbędnym źródłem zaopatrzenia dla wszystkich komórek, które mogą być dostarczane tylko przez mitochondria.
Choroby
Mitochondriopatie, nieprawidłowe działanie lub zaburzenia mitochondrialnych procesów metabolicznych wpływają przede wszystkim na osłabienie wydolności organizmu ze względu na zmniejszoną syntezę ATP.
Zasadniczo choroba mitochondrialna może być spowodowana odziedziczonymi wadami genetycznymi lub została nabyta w toku życia. Nabyte choroby mitochondrialne są związane przede wszystkim z chorobami neurodegeneracyjnymi, takimi jak choroba Alzheimera, choroba Parkinsona i ALS, ale także z cukrzycą, otyłością, chorobami układu krążenia i niektórymi rodzajami raka.
Dziedziczne choroby mitochondrialne wykazują różnorodne objawy, w zależności od tego, gdzie w kaskadzie metabolicznej wpływa defekt genetyczny. Jeśli z powodu defektów genetycznych w łańcuchu oddechowym lub cyklu kwasu cytrynowego nie są dostępne pewne enzymy, które są wymagane tylko dla określonych tkanek ciała, objawy występują tylko w odpowiednich narządach.
Ze względu na różnorodność objawów, które może powodować defekt genów mitochondrialnych, diagnoza nie jest łatwa. Ponieważ zwykle wpływa to również na cykl kwasu cytrynowego, pirowian „gromadzi się”, który organizm próbuje rozłożyć drogą alternatywną do mleczanu, w wyniku czego dochodzi do znacznie zwiększonego stężenia kwasu mlekowego, czyli kwasicy mleczanowej.

.jpg)
.jpg)


.jpg)
.jpg)