Plik dystrofia miotoniczna typu 1 (zespół Curschmanna-Steinerta) jest autosomalnie dominującą, dziedziczną chorobą wieloukładową z głównymi objawami osłabienia mięśni i zmętnienia soczewki (zaćma). Rozróżnia się dwie formy choroby: postać wrodzoną (wrodzona), w której noworodek jest zauważalny z powodu osłabienia mięśni („wiotkie niemowlę”) oraz postać dorosłą, która objawia się dopiero w drugiej i trzeciej dekadzie życia. Dystrofia miotoniczna typu 1 jest nieuleczalna iw zależności od jej nasilenia i progresji skraca oczekiwaną długość życia.
Co to jest dystrofia miotoniczna typu 1?
© peshkova - stock.adobe.com
Plik dystrofia miotoniczna typu 1 jest jedną z tak zwanych chorób powtórzeń trinukleotydów. W kodzie genetycznym na długim ramieniu chromosomu 19 zduplikowany jest trinukleotyd z zasad nukleinowych cytozyny, tyminy i guaniny.
Podczas gdy ta podstawowa trójka powtarza się 5-35 razy u zdrowych osób, u osób z łagodnymi objawami jest to około 50-200, w ciężkich formach nawet ponad 1000 powtórzeń. Trinukleotyd nie koduje bezpośrednio białka, ale wpływa na syntezę innych białek. Enzym niezbędny w mięśniach szkieletowych i sercowych, kinaza białkowa dystrophia myotonica (DMPK), jest wytwarzana w ograniczonym stopniu z powodu defektu genetycznego.
Ale wpływa to również na inne białka, np. SIX5 wyrażany w soczewce lub receptorze insuliny. Dlatego dystrofia miotoniczna typu 1 wpływa na wiele różnych układów narządów. Dystrofia miotoniczna typu 1, występująca z częstością około 1:20 000, jest najczęstszą miotonią i jednocześnie najczęstszą dystrofią mięśniową występującą w wieku dorosłym.
Pod względem dziedziczenia liczba powtórzeń trinukleotydów wzrasta z pokolenia na pokolenie, tak że początek choroby u potomstwa jest wcześniejszy i cięższy. Forma wrodzona jest zawsze dziedziczona po matce. Dystrofia miotoniczna typu 1 częściej dotyka chłopców niż dziewczynki.
przyczyny
W postaci wrodzonej niemowlę jest widoczne zaraz po urodzeniu z uogólnionym osłabieniem mięśni, podniesioną górną wargą i niewydolnością oddechową. Z powodu problemów z oddychaniem wiele noworodków jest uzależnionych od sztucznego oddychania, a 25% -50% umiera w ciągu pierwszych 18 miesięcy życia.
U dzieci, które przeżywają dłużej, należy się spodziewać opóźnień rozwojowych i poważnego upośledzenia umysłowego. Ich średnia długość życia wynosi około 30-40 lat. Jeśli choroba ujawni się dopiero w wieku dorosłym, osoby dotknięte chorobą często najpierw zauważają osłabienie mięśni odległych od tułowia, zwłaszcza nóg, szyi i twarzy. Zanik mięśni twarzy sprawia, że pacjent wygląda na wychudzonego.
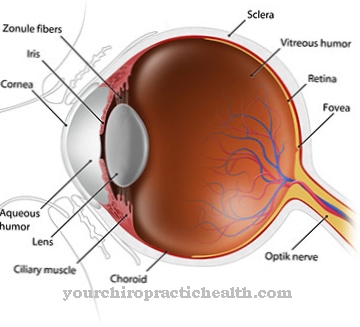
Powodują również zaburzenia mowy i połykania. Osłabieniu mięśni towarzyszy opóźnione rozluźnienie mięśni, tak że osobom dotkniętym chorobą trudno jest na przykład ponownie rozluźnić ruch. Dalsze objawy to zmętnienie soczewki, utrata słuchu w uchu wewnętrznym, zwiększona potrzeba snu, ograniczenia poznawcze i zmniejszona tolerancja glukozy, aż do cukrzycy. Z powodu zaburzonej równowagi hormonalnej zanik jąder i łysienie występują zwykle u mężczyzn, a zaburzenia miesiączkowania u kobiet.
Szczególnie niebezpieczny jest wpływ na mięśnie serca: często występują zaburzenia rytmu serca, a niekiedy nawet zatrzymanie akcji serca. Jeśli osłabienie mięśni dotrze do mięśni rdzeniowych, skutkuje to zaburzeniami oddychania i zwiększoną podatnością na infekcje płuc. Dystrofia miotoniczna typu 1 zawsze postępuje, ale nasilenie i skład jej objawów są niezwykle zmienne. Średnia długość życia dorosłej postaci dystrofii miotonicznej typu 1 wynosi około 50-60 lat.
Objawy, dolegliwości i oznaki
Główną cechą dystrofii miotonicznej typu 1 jest opóźnione rozluźnienie mięśni po ich skurczu. Ta cecha może służyć do odróżnienia choroby od innych dystrofii mięśniowych. Szczególnie dotknięte są mięśnie najbardziej oddalone od tułowia, takie jak mięśnie twarzy, szyi, przedramion, dłoni, podudzi i stóp. Istnieją inne objawy niezależne od dolegliwości mięśni.
Często występują zaburzenia rytmu serca lub niewydolność serca. Ze względu na zajęcie serca podczas znieczulenia często dochodzi do incydentów znieczulenia. Często obserwuje się zaćmę i łysienie włosów u mężczyzn. Poziom testosteronu jest zbyt niski, co często prowadzi do utraty jąder. Istnieje zwiększone ryzyko rozwoju cukrzycy.
Mówienie i połykanie są trudne dla pacjenta. Ponadto pacjent jest ciągle zmęczony w ciągu dnia, co może prowadzić do nocnych przerw w oddychaniu. Jednak bezdech senny nie zawsze występuje. Jako dalsze objawy mogą również wystąpić zaburzenia trawienia, zaburzenia woreczka żółciowego lub zaburzenia słuchu. Chociaż jest to choroba dziedziczna, objawy pojawiają się u wielu pacjentów dopiero w wieku 20 lat.
Zaćma jest często rozpoznawana jako pierwszy objaw choroby.Jednak istnieje również forma choroby, która istnieje od urodzenia. Ta wrodzona postać miotonii mięśniowej charakteryzuje się szczególnie ciężkim przebiegiem z zagrażającą życiu niewydolnością oddechową oraz zaburzeniami rozwoju psychicznego i fizycznego.
Diagnoza i przebieg
Jeśli podejrzewasz dystrofia miotoniczna typu 1 Do bezsprzecznego rozpoznania choroby wykorzystuje się molekularne metody genetyczne. Pomagają one wykluczyć diagnostykę różnicową z podobnymi objawami, np. Dystrofia miotoniczna typu 2. Rozpoznanie może być poparte badaniami elektromiograficznymi (EMG). U osób dotkniętych chorobą można znaleźć typowe wzorce spontanicznej aktywności, zwłaszcza na mięśniach odległych od tułowia. Ważne jest również, aby mieć dokładną historię rodzinną, również w celu udzielenia dalszych porad rodzinie.
Komplikacje
W przypadku tej choroby osoby dotknięte chorobą cierpią głównie na poważne osłabienie mięśni i objawy występujące w oczach. Prowadzi to do zaćmy i zmętnienia soczewki, przez co wzrok chorego znacznie się pogarsza. W najgorszym przypadku może również doprowadzić do całkowitej ślepoty.
Jakość życia jest znacznie obniżona. Szczególnie młodzi ludzie mogą mieć dolegliwości psychiczne lub depresję, jeśli mają nagłe problemy ze wzrokiem lub są niewidomi. Ponadto mogą wystąpić problemy z sercem, tak że pacjent może umrzeć z powodu nagłej śmierci sercowej. Nierzadko zdarza się, że osoby dotknięte chorobą chorują na cukrzycę.
Osłabienie mięśni znacznie ogranicza codzienne życie osób dotkniętych chorobą, przez co w niektórych przypadkach są również zależni od pomocy innych osób. Pewnych zajęć lub sportów nie można już uprawiać bez dalszych ceregieli. Choroba znacznie ogranicza rozwój dzieci, przez co w wieku dorosłym mogą wystąpić komplikacje. Nie ma możliwości leczenia przyczynowego tej choroby.
Jednak wiele skarg można ograniczyć i złagodzić, tak aby życie codzienne osoby zainteresowanej stało się znośne. Z reguły nie występują żadne szczególne powikłania, a oczekiwana długość życia pacjenta nie jest ograniczona chorobą.
Kiedy powinieneś iść do lekarza?
Wizyta u lekarza jest konieczna, gdy osoba zainteresowana doświadcza trudności w radzeniu sobie w życiu codziennym. Osłabienie siły mięśni, spadek wydolności fizycznej i utrata tkanki to oznaki zaburzeń zdrowia. Jeśli zwykłe zajęcia sportowe można uprawiać tylko w ograniczonym zakresie lub nie można ich wcale uprawiać, obserwacje należy omówić z lekarzem. Konieczne jest rozpoczęcie różnych badań, aby wyjaśnić przyczynę i sporządzić plan leczenia.
Niepokojące jest opóźnienie w dobrowolnie kontrolowanym napięciu mięśni i pogorszenie wzroku. Jeśli widzisz niewyraźne lub mętne soczewki, wskazana jest wizyta lekarska. Nieregularność w naturalnej funkcji chwytu jest sygnałem ostrzegawczym od ciała, który wymaga działania. Zwiększone ryzyko wypadków i upadków należy omówić z lekarzem, aby można było zastosować środki zaradcze. Zaburzenia rytmu serca, kołatanie serca lub zaburzenia snu powinny zostać dokładniej zbadane przez lekarza.
W przypadku wystąpienia zaburzeń koncentracji, uwagi lub obniżenia sprawności umysłowej z powodu upośledzenia konieczna jest wizyta lekarska. Jeśli mężczyźni cierpią na zmniejszone pożądanie seksualne lub jeśli mają łysinę, należy skonsultować się z lekarzem. Jeśli występują również stany stresu emocjonalnego lub psychicznego, osobie dotkniętej chorobą grożą konsekwencje. Należy im zapobiegać w odpowiednim czasie.
Leczenie i terapia
Leczenie przyczynowe dystrofia miotoniczna typu 1 Nie mogę. Terapia koncentruje się na łagodzeniu objawów, np. poprzez chirurgiczne leczenie zaćmy, dostosowanie leków przy zaburzeniach rytmu serca czy techniczne wsparcie oddechowe. Wsparcie fizjoterapeutyczne może opóźnić postęp dystrofii miotonicznej typu 1.
Tutaj znajdziesz swoje leki
➔ Leki na osłabienie mięśniPerspektywy i prognozy
Perspektywy rozpoznania dystrofii miotonicznej typu 1 są słabe. Cierpi na tym zarówno oczekiwana długość życia, jak i jakość życia. Większość pacjentów nie osiąga nawet wieku 60 lat. Wielu z nich umiera z powodu niewydolności serca lub ulega infekcjom. Środki terapeutyczne mogą często tylko marginalnie złagodzić objawy choroby. Zgodnie z obecnym stanem nauki, sama wada genetyczna nie jest uleczalna. Wiele osób dotkniętych chorobą wykazuje oznaki dystrofii miotonicznej typu 1. przed ukończeniem 20. roku życia. Inni odwiedzają lekarza dopiero w podeszłym wieku. W rodzinach istnieje zwiększone ryzyko dziedziczenia choroby.
Cierpienie narasta w sposób ciągły, ponieważ dystrofia miotoniczna typu 1 postępuje nieustannie przez lata. Z powodu słabych mięśni osobom dotkniętym chorobą coraz trudniej jest radzić sobie w pojedynkę w codziennym życiu. Potrzebujesz pomocy. Układ mięśniowo-szkieletowy zatrzymuje się. Po jakimś czasie nie można już wykonywać wyuczonego zawodu. Podejścia terapeutyczne leków i fizjoterapii z czasem coraz bardziej tracą na skuteczności. Nierzadko fizycznemu zanikowi dystrofii miotonicznej typu 1 towarzyszą problemy psychologiczne.
zapobieganie
Ponieważ dystrofia miotoniczna typu 1 Jeśli jest to dziedziczna wada genetyczna, zapobieganie nie jest możliwe.
Opieka postpenitencjarna
Dystrofia miotoniczna typu 1 jest dziedziczna. Zgodnie z obecnym stanem badań wyleczenie nie jest możliwe. Choroba skraca oczekiwaną długość życia o około 50 lat. Wskazana jest dalsza opieka, aby spowolnić postęp dystrofii. Dalsze cele opieki pooperacyjnej to złagodzenie objawów i utrzymanie jakości życia.
Podczas obserwacji sprawdza się tolerancję leku, czy został on podany pacjentowi. Opieka kontrolna dotyczy przede wszystkim dolegliwości fizycznych. Należy jak najdłużej utrzymywać ruchomość kończyn poprzez odpowiednie ćwiczenia. Towarzysząca psychoterapia może być również odpowiednia lub nawet konieczna.
Niedostateczna jakość życia spowodowana dystrofią może wpływać na duszę pacjenta. Ryzyko depresji jest bardzo wysokie. Dzięki psychoterapii istnieje możliwość porozmawiania o negatywnych uczuciach. Na zaawansowanym etapie może być wymagany wózek inwalidzki. W trakcie opieki pooperacyjnej chory uczy się codziennej obsługi urządzenia.
Dystrofia miotoniczna wpływa również na czynność serca. Stymulator przeciwdziała temu procesowi. Dalsze leczenie przeprowadza kardiolog. Monitoruje proces gojenia po operacji. Kontrola zostanie przerwana, gdy leczenie przebiegnie zgodnie z oczekiwaniami.
.jpg)






.jpg)


.jpg)