Plik Biosynteza cholesterolu umożliwia komórkom organizmu syntezę cholesterolu z prostych surowców w 18 etapach. Ta biosynteza zachodzi głównie w wątrobie i ścianach jelit. Dziedziczne choroby metaboliczne, takie jak choroba Tangeru, mogą zakłócać biosyntezę cholesterolu.
Czym jest biosynteza cholesterolu?

Organizm ludzki wytwarza swój własny cholesterol w procesie biochemicznym w 18 różnych etapach. Ten proces jest również znany jako biosynteza cholesterolu. Większość cholesterolu całkowitego jest wytwarzana przez organizm. Tylko minimalna frakcja jest wchłaniana z pożywieniem.
Cholesterol to lipid, który jest niezbędny dla wielu funkcji organizmu. Na przykład w przypadku syntezy hormonów steroidowych organizm jest zależny od cholesterolu. To samo dotyczy różnych procesów przechowywania i budowy błon komórkowych.
Szlak metaboliczny biosyntezy cholesterolu umożliwia wszystkim żywym istotom posiadającym jądro komórkowe wytwarzanie ważnych lipidów z prostych elementów. W razie potrzeby reguluje się produkcję cholesterolu w organizmie. Transformacja substancji zachodzi w cytozolu i retikulum endoplazmatycznym komórek. Czynniki transkrypcyjne regulują procesy i wpływają pozytywnie lub negatywnie na biosyntezę.
Bloch i Lynen otrzymali Nagrodę Nobla w 1964 roku za badania nad metabolizmem cholesterolu. Popják i Cornforth również wnieśli ważny wkład w badania nad biosyntezą cholesterolu.
Funkcja i zadanie
Ludzkie ciało wytwarza codziennie około 700 miligramów cholesterolu w procesie biosyntezy, a około 150 gramów cholesterolu jest wbudowywane w całe ciało. Duże ilości lipidów znajdują się głównie w mózgu i nadnerczach. Cholesterol pełni funkcje stabilizujące w błonach komórkowych i dlatego jest ważnym budulcem.
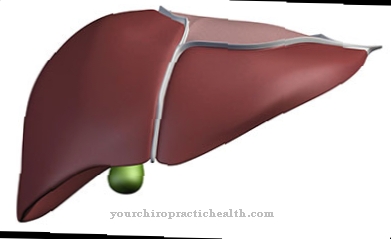
U ludzi biosynteza cholesterolu zachodzi przede wszystkim w błonie śluzowej jelit i wątrobie. Chociaż wiele komórek organizmu jest zdolnych do syntezy cholesterolu, wątroba nadal wytwarza większość cholesterolu.
Ponieważ cholesterol w organizmie nie może przekroczyć bariery krew-mózg do mózgu, mózg musi sam wytwarzać cholesterol w ośrodkowym układzie nerwowym. Cholesterol w mózgu stanowi około 24 procent całkowitego cholesterolu.
Produktem syntezy cholesterolu jest DMAPP, który jest wytwarzany na szlaku metabolicznym mewalonianu. 18 etapów pośrednich składa się na biosyntezę cholesterolu. Przed syntezą organizm syntetyzuje acetylo-CoA. Proces ten zachodzi na szlaku biosyntezy mewalonianu. Poprzez HMG-CoA, substrat acetylo-CoA przekształca się w kwas mewalonowy. Końcowymi produktami szlaku biosyntezy mewalonianu są pirofosforan dimetyloallilu i pirofosforan izopentylu.
Dopiero teraz zaczyna się właściwa biosynteza cholesterolu. Dwa produkty końcowe szlaku biosyntezy meyalonianu łączą się, tworząc pirofosforan geranylu. Ten związek jest przekształcany w pirofosforan farnezylu. W reakcji kondensacji biorą udział dwa pirofosforany farnezylu, które w trakcie tej reakcji przekształcają się w skwalen. Z tego powstaje (S) -2,3-epoksyskwalen, który z kolei przekształca się w lanosterol. Lanosterol bierze udział w demetylacji. Staje się więc 4,4-dimetylo-5α-cholesta-8,14,24-trien-3β-ol.
W tym momencie zachodzi kilka reakcji utleniania, które prowadzą do powstania 14-demetyllanosterolu. Końcowe produkty utleniania są przekształcane w zymosteron poprzez karboksylan zymosterolu. Następnie następuje redukcja zymosteronu, co skutkuje zymosterolem. Poprzez 5α-Cholesta-7,24-dien-3β-ol prowadzi to do 7-dehydrocholesterolu. Kiedy ten produkt jest uwodorniony, ostatecznie powstaje cholesterol.
Choroby i dolegliwości
Różne choroby dziedziczne, które wpływają na metabolizm cholesterolu, znane są pod nazwą rodzinnej hipercholesterolemii. Niezależnie od diety, zaburzenia te powodują znaczny wzrost poziomu cholesterolu w osoczu. Choroby naczyniowe i zawały serca mogą pojawić się we wczesnym wieku jako choroby wtórne. Choroba jest spowodowana defektem genu kodującego receptor LDL. Z powodu tej wady receptor LDL jest tylko niecałkowicie rozwinięty lub wcale. Przede wszystkim zatem wartość LDL osób dotkniętych chorobą jest znacznie zwiększona. Ksantomy kształtują obraz kliniczny. Są to złogi tłuszczu w skórze, w narządach wewnętrznych oraz w ośrodkowym układzie nerwowym.
Hipercholesterolemia nie musi być rodzinna, można ją też nabyć. Nabyte formy są głównie wywoływane przez niedożywienie. Cukrzyca może być chorobą podstawową. Otyłość lub przewlekła niewydolność nerek są również często związane ze zwiększonym poziomem cholesterolu.
Oprócz diet leki, takie jak inhibitory CSE, są głównie stosowane w leczeniu ponadprzeciętnie wysokiego poziomu cholesterolu. Statyny mogą hamować biosyntezę cholesterolu. Leczenie inhibitorami CSE ma na celu to zahamowanie. Zapobiegają reduktazie HMG-CoA, a tym samym umożliwiają ogólne obniżenie stężenia cholesterolu w surowicy. W ten sposób można na przykład spowolnić wtórną chorobę miażdżycową. Zawałom serca związanym z cholesterolem lub innym współistniejącym chorobom, w których poziom cholesterolu jest zbyt wysoki, można również zapobiec za pomocą inhibitora CSE.
Hipocholesterolemie są przeciwieństwem hipercholesterolemii. Niski poziom cholesterolu w surowicy, który występuje w kontekście hipocholesterolemii, jest w większości przypadków związany z rakiem złośliwym. W hipocholesterolemii związanej z rakiem niski poziom cholesterolu jest zwykle oceniany jako czynnik ryzyka zgonu z jakiejkolwiek przyczyny.
Niedożywienie, AIDS lub ciężkie infekcje to inne przyczyny bardzo niskiego poziomu cholesterolu. Jednak hipocholesterolemia może również wystąpić jako część choroby dziedzicznej. Przykładem takiej choroby jest choroba Tangeru. Pacjenci z tą chorobą szczególnie cierpią na hipocholesterolemię HDL.





.jpg)
.jpg)


.jpg)
.jpg)