Plik Choroba magazynowania kwasu sialowego jest bardzo rzadko występującą, genetyczną lizosomalną chorobą spichrzeniową, w której białko sialina jest nieprawidłowo kodowane. Sialina jest białkiem transbłonowym, które normalnie usuwa monosacharydy, takie jak kwas sialowy, które powstają w wyniku enzymatycznego rozkładu glikoprotein i innych substancji z lizosomu. Zmniejszona funkcjonalność sialiny prowadzi do gromadzenia się kwasu sialowego w lizosomach, czyli do choroby spichrzeniowej kwasu sialowego.
Co to jest choroba spichrzeniowa kwasu sialowego?

© LIGHTFIELD STUDIOS - stock.adobe.com
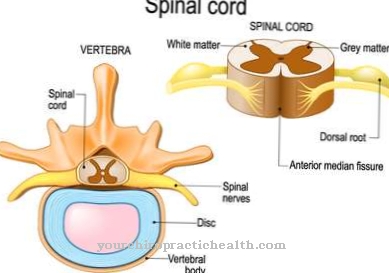
Termin kwas sialowy obejmuje pochodne kwasu neuraminowego, które są N- lub O-acetylowane. Kwasy sialowe są budulcem glikoprotein, glikozaminoglikanów i innych substancji i są uwalniane w wyniku ich enzymatycznego rozpadu w specjalnych lizosomach.
Zwykle kwasy sialowe są odprowadzane z lizosomu przez błonę lizosomu za pomocą sialiny, białka transportującego aniony. Charakterystyka Choroba magazynowania kwasu sialowego, które są również nazywane Choroba spichrzeniowa kwasu neuraminowego jest funkcjonalnym ograniczeniem lub całkowitą utratą funkcji transportującego białka sialiny.
Kwasy sialowe wytwarzane w lizosomach mogą być odprowadzane z lizosomów, tak że dochodzi do nadmiernej akumulacji kwasów sialowych w lizosomach. Znane są dziecięce i dorosłe formy lizosomalnej choroby spichrzeniowej kwasu sialowego. Postać dorosła jest również znana jako choroba Salli.
przyczyny
Przyczyny utraty funkcji transbłonowego białka sialiny sięgają defektu lub mutacji genu SLC17A5 zarówno u postaci niemowlęcej, jak i dorosłej. Gen znajduje się na chromosomie 6, w locus genu q14-q15. Wszystkie znane mutacje genu prowadzą do nieprawidłowego kodowania białka sialiny.
Chociaż jest akceptowany jako budulec w metabolizmie, nie ma zdolności uwalniania kwasu sialowego przez błonę lizosomu. Niewłaściwe kodowanie sialiny nadaje białku inną strukturę trzeciorzędową, która jest związana z utratą funkcjonalności białka transportowego. Różne mutacje genów prowadzą do infantylnej lub dorosłej postaci choroby spichrzeniowej kwasu sialowego.
Objawy, dolegliwości i oznaki
Niemowlęca choroba spichrzeniowa kwasu sialowego jest chorobą o mniej korzystnym rokowaniu dla obu postaci. Pierwsze oznaki i objawy pojawiają się przed urodzeniem. Diagnostyka prenatalna ultrasonograficzna zwykle ujawnia tak zwane obrzęk płodu, nagromadzenie płynu tkankowego, który obejmuje duże części płodu.
Po urodzeniu powiększenie wątroby i śledziony jest objawowe, a rysy twarzy są przeważnie szorstkie. Oczekiwana długość życia w infantylnej postaci choroby spichrzeniowej kwasu neuraminowego wynosi zwykle tylko kilka lat. Choroba Salla lub dorosła postać choroby spichrzeniowej kwasu sialowego jest już zauważalna w okresie niemowlęcym.
Dzieje się to poprzez niedociśnienie mięśniowe i oczopląs poziomy, w którym oczy wykonują szarpane ruchy poziome. Osoby dotknięte chorobą są upośledzone umysłowo i najczęściej nie mają zdolności językowych. Prognozy dotyczące oczekiwanej długości życia są nieco bardziej korzystne niż prognozy dotyczące dziecięcej postaci choroby. Osoby dotknięte chorobą zwykle osiągają wiek dorosły.
Diagnoza i przebieg choroby
U osób dotkniętych chorobą spichrzeniową kwasu sialowego objawowe jest zwiększone stężenie kwasu sialowego w moczu i zwiększona szybkość wydalania polisacharydów. Wolny kwas sialowy można wykryć w fibroblastach i komórkach owodniowych. Ostateczną pewnością jest badanie genetyczne, które można przeprowadzić również w okresie prenatalnym.
W ciężkich przypadkach choroby spichrzeniowej kwasu sialowego, wady rozwojowe kości, wodobrzusze (wodobrzusze), ciężkie zaburzenia ruchowe, skurcze spastyczne i poważne upośledzenie umysłowe stają się zauważalne wkrótce po urodzeniu. Ciężkie postacie choroby prowadzą do śmierci we wczesnym dzieciństwie.
Przy umiarkowanych przebiegach - typowych dla choroby Salli - osoby dotknięte chorobą zwykle osiągają wiek dorosły. Ciężka niepełnosprawność intelektualna, konwulsje spastyczne i poważne ograniczenia psychomotoryczne są objawami tych postaci choroby. Chociaż choroba jest bardzo rzadka na całym świecie, jej ognisko regionalne występuje w północnej Finlandii, gdzie co 40 heterozygotyczny nosiciel jest jedną z odpowiadających im mutacji genów.
Komplikacje
W większości przypadków chorobę spichrzeniową kwasu sialowego można rozpoznać i zdiagnozować przed urodzeniem. Osoby dotknięte chorobą zwykle cierpią na znacznie powiększoną śledzionę i wątrobę. Może to prowadzić do bólu w odpowiednich regionach. Rysy twarzy pacjentów są często szorstkie, więc może wystąpić dokuczanie lub zastraszanie, szczególnie w młodym wieku.
Wiele dzieci cierpi na dolegliwości psychiczne lub depresję. Ponadto osoby dotknięte chorobą często cierpią na niepełnosprawność intelektualną, a tym samym na ograniczenia w życiu codziennym. Dlatego często są zależni od pomocy innych ludzi w życiu codziennym i nie mogą z łatwością wykonywać wielu codziennych czynności. Choroba spichrzeniowa kwasu sialowego może również prowadzić do problemów językowych, tak że komunikacja między osobami dotkniętymi chorobą jest znacznie ograniczona.
Ze względu na chorobę spichrzeniową kwasu sialowego oczekiwana długość życia pacjenta jest zwykle również znacznie zmniejszona. Większość osób dotkniętych chorobą osiąga z tym dopiero dorosłość. Ponieważ leczenie przyczynowe nie jest możliwe, można ograniczyć tylko objawy. Nie ma jednak szczególnych komplikacji. Nie można zapobiec chorobie spichrzeniowej kwasu sialowego.
Kiedy powinieneś iść do lekarza?
Choroba spichrzeniowa kwasu sialowego zwykle zawsze wymaga badania lekarskiego i leczenia. Ponieważ jest to choroba dziedziczna, w przypadku chęci posiadania dzieci należy również przeprowadzić badanie genetyczne, aby zapobiec przenoszeniu choroby na potomstwo. Ponieważ nie prowadzi to do samoleczenia, osoba dotknięta chorobą spichrzeniową kwasu sialowego jest w każdym przypadku zależna od leczenia przez całe życie.
W przypadku znacznego powiększenia śledziony lub wątroby należy skonsultować się z lekarzem. W większości przypadków objawy te są wykrywane przypadkowo podczas badania ultrasonograficznego, chociaż ból po bokach ciała może również wskazywać na te objawy. W niektórych przypadkach problemy z mówieniem lub niektóre niepełnosprawności intelektualne wskazują na chorobę spichrzeniową kwasu sialowego.
Pierwsze badanie tej skargi może przeprowadzić lekarz rodzinny. Dalsze leczenie jest zwykle przeprowadzane przez różnych specjalistów i zawsze opiera się na dokładnych objawach.
Leczenie i terapia
Ponieważ choroba spichrzeniowa kwasu sialowego ma podłoże genetyczne, leczenie przyczynowe nie jest możliwe. Nie ma sposobu, aby zastąpić funkcjonalnie upośledzone białko transbłonowe i transportujące aniony funkcjonalną sialiną. Możliwe terapie są (nadal) ograniczone do leczenia objawów i zapewnienia, aby pacjent odczuwał jak najmniej bólu.
W przypadku niektórych innych lizosomalnych chorób spichrzeniowych, takich jak choroba Huntera i choroba Gauchera, infuzja genetycznie zmodyfikowanych enzymów już przyniosła pozytywne rezultaty. Procedura ta jest nazywana enzymatyczną terapią zastępczą i jest skuteczna w przypadku narządów wewnętrznych i tkanki łącznej. Ze względu na swoją wielkość sztucznie dostarczane enzymy nie mogą przekroczyć bariery krew-mózg, więc takie terapie nie mają wpływu na rozwój OUN.
Jedną z form terapii, która jest obecnie jeszcze w powijakach, jest tak zwana terapia genowa, która ma doprowadzić do ukierunkowanej wymiany zmutowanej sekwencji genów. Zasadniczo jest to technika, która jest stosowana w pozytywnym sensie do naprawy własnych genów organizmu lub może mieć również negatywne skutki w postaci pewnych patogennych zarazków.
Tutaj znajdziesz swoje leki
➔ Leki przeciwbólowezapobieganie
Nie ma środków zapobiegawczych, które mogłyby zapobiec wybuchowi choroby spichrzeniowej kwasu sialowego, ponieważ jest to genetycznie uwarunkowana, ogólnoustrojowo skuteczna choroba, która powstała już w okresie prenatalnym.
W rodzinach, w których na przykład znane są przypadki choroby Salli, jeśli chcą mieć dzieci, można przeprowadzić badanie genetyczne w celu ustalenia, czy potencjalny rodzic jest nosicielem jednej ze znanych mutacji genów, nie będąc sam chorym. W pozytywnym przypadku powinna odbyć się konsultacja, podczas której należy wyjaśnić ryzyko, jakie istnieje dla dziecka w realizacji pragnienia posiadania dzieci.
Opieka postpenitencjarna
Z reguły pacjenci z chorobą spichrzeniową kwasu sialowego nie mają żadnych specjalnych możliwości bezpośredniej opieki kontrolnej. Z tego powodu choroba ta powinna być wcześnie diagnozowana i leczona przez lekarza, aby w dalszym przebiegu nie wystąpiły powikłania i inne objawy.
Im wcześniej konsultacja z lekarzem, tym zwykle lepszy jest dalszy przebieg choroby, dlatego należy zgłosić się do lekarza, gdy tylko pojawią się pierwsze objawy. Choroba spichrzeniowa kwasu sialowego zwykle nie może wyleczyć się sama. Większość pacjentów z chorobą spichrzeniową kwasu sialowego jest uzależniona od przyjmowania różnych leków w celu trwałego i prawidłowego złagodzenia objawów choroby.
W celu prawidłowego przeciwdziałania objawom należy przestrzegać regularnego spożycia i przepisanej dawki. Bardzo ważne są również regularne wizyty kontrolne u lekarza w celu wczesnego rozpoznania i leczenia innych uszkodzeń. Z powodu choroby część osób dotkniętych chorobą jest również zależna od pomocy i wsparcia własnych rodzin. Ta choroba może również skrócić oczekiwaną długość życia pacjenta.
Możesz to zrobić sam
Ponieważ choroba jest spowodowana defektem genetycznym, osoba dotknięta chorobą nie może wybierać opcji, które przyczyniają się do wyleczenia zaburzenia zdrowia. Środki samopomocy powinny być skoncentrowane na poprawie codziennych procesów i poprawie samopoczucia.
Należy wziąć pod uwagę, że duża liczba pacjentów cierpi na zaburzenia funkcji poznawczych. Dlatego często są zależni od pomocy i wsparcia krewnych lub personelu pielęgniarskiego wyszkolonego medycznie. Zmagając się z chorobą, szczególnie rodzice lub inni członkowie rodziny muszą brać pod uwagę własne ograniczenia i nie rezygnować z opieki i wsparcia. Rozwój elementów poprawiających radość życia jest niezmiernie ważny dla wszystkich zaangażowanych. Stabilne środowisko społeczne pomaga również w przeciwnościach.
Ponieważ choroba ma negatywny wpływ na rozwój niektórych narządów, należy to wziąć pod uwagę w życiu codziennym. Zdrowa dieta, optymalna higiena snu i regularne codzienne czynności pomagają radzić sobie z chorobą. Zapewniają bezpieczeństwo i stabilność. Ponieważ komunikacja z pacjentem jest ograniczona, należy rozwinąć możliwości, aby w życiu codziennym miała miejsce dobra wymiana wszystkich zaangażowanych osób. Wzajemne zrozumienie, spokój i unikanie gorączkowego tempa sprzyjają dalszemu rozwojowi.

.jpg)




.jpg)


.jpg)