Plik Lipaza lipoproteinowa (LPL) należy do lipaz i odgrywa kluczową rolę w metabolizmie lipidów. Odpowiada za rozszczepianie trójglicerydów w chylomikronach i lipoproteinach o bardzo małej gęstości (VLDL) na kwasy tłuszczowe i monoacyloglicerynę. Uwolnione kwasy tłuszczowe są wykorzystywane do wytwarzania energii lub do budowy tkanki tłuszczowej.
Co to jest lipaza lipoproteinowa?
Lipaza lipoproteinowa (LPL) to enzym będący jedną z lipaz. Lipazy są odpowiedzialne za rozkład trójglicerydów (triacylogliceroli) na kwasy tłuszczowe i glicerynę. Trójglicerydy to estry trójalkoholu gliceryny, z których każdy zawiera trzy kwasy tłuszczowe, znane jako tłuszcze lub oleje tłuszczowe.
Tłuszcze pokarmowe są wchłaniane z pożywieniem i najpierw są rozkładane przez zewnątrzkomórkowe lipazy z trzustki w jelicie. Jednak niektóre trójglicerydy przedostają się do krwiobiegu poprzez wchłanianie w jelicie cienkim przez surowicę, gdzie są związane z lipoproteinami, które gwarantują ich transport we krwi. Lipaza lipoproteinowa jest enzymem, który rozkłada trójglicerydy związane z lipoproteinami na kwasy tłuszczowe i monoacyloglicerol. Składa się z 448 aminokwasów i jest zależny od koenzymu apolipoproteiny C2.
Lipaza lipoproteinowa jest rozpuszczalnym w wodzie enzymem, który jest związany z komórkami śródbłonka naczyń krwionośnych poprzez określone glikoproteiny (proteoglikany). Jest produkowany w wątrobie. Enzym katalizuje hydrolizę trójglicerydów, tworząc dwie cząsteczki kwasów tłuszczowych i jedną cząsteczkę monoacyloglicerolu. Apolipoproteiny są cząsteczkami nośnikowymi trigliceryn i umożliwiają ich transport w środowisku wodnym. Apolipoproteina C2 działa również jako receptor lipazy lipoproteinowej, a tym samym aktywuje hydrolizę trójglicerydów.
Funkcja, efekt i zadania
Funkcją lipazy lipoproteinowej jest całkowite katalizowanie rozpadu we krwi tłuszczów wchłanianych przez komórki jelitowe. Po pierwsze, tłuszcze zawarte w diecie są rozkładane na kwasy tłuszczowe i glicerynę przez lipazy trzustkowe w jelicie cienkim. Dalsze trójglicerydy dostają się do krwi poprzez wchłanianie przez jelito cienkie i tam wiążą się z lipoproteinami, tworząc kompleks lipidowo-białkowy.
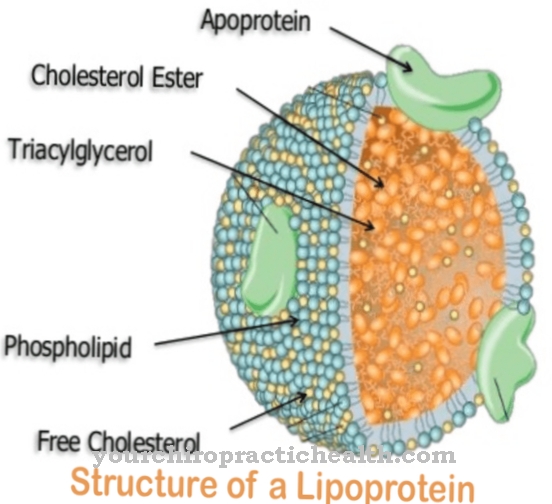
To tworzy chylomikrony. Reprezentują cząsteczki lipoprotein o średnicy od 0,5 do 1 mikrometra, a ich gęstość nie przekracza 1000 g / ml. Rdzeń lipidowy zawiera głównie trójglicerydy z niewielką ilością estrów cholesterolu. Powłoka chylomikronów zawierająca cholesterol zawiera fosfolipidy jako element strukturalny. Apolipoproteiny, z którymi związane są trójglicerydy, są teraz również przechowywane w tej powłoce. Chylomikrony zawierają 90 procent trójglicerydów. Dostają się do krwiobiegu z jelita cienkiego przez układ limfatyczny. Trójglicerydy są rozkładane na kwasy tłuszczowe i glicerynę za pomocą LPL, szczególnie w naczyniach włosowatych tkanki mięśniowej i tłuszczowej.
Kwasy tłuszczowe są wykorzystywane w tkance mięśniowej do wytwarzania energii lub w tkance tłuszczowej do budowy endogennych trójglicerydów jako tłuszczu zapasowego. Po około dziesięciu godzinach abstynencji pokarmowej nie można już wykryć chylomikronów we krwi, ponieważ trójglicerydy są wtedy całkowicie rozkładane. Inne składniki krwi to tak zwane VLDL (lipoproteiny o bardzo małej gęstości). Te jednostki strukturalne są uwalniane z wątroby i zawierają trójglicerydy, fosfolipidy i cholesterol. VLDL transportuje te składniki przez krwioobieg z wątroby do poszczególnych narządów.
W ten sposób trójglicerydy są rozkładane przez lipazę lipoproteinową, a uwolnione kwasy tłuszczowe są wchłaniane przez komórki organizmu. Spadek trójglicerydów przekształca VLDL w LDL (lipoproteiny o niskiej gęstości). LDL zawiera głównie fosfolipidy, estry cholesterolu i lipoproteiny
Edukacja, występowanie, właściwości i optymalne wartości
Lipaza lipoproteinowa jest syntetyzowana w wątrobie. Oprócz lipaz trzustkowych reprezentuje inną lipazę zewnątrzkomórkową LPL znajduje się na zewnątrz błon komórek śródbłonka różnych narządów, w tym komórek tłuszczowych. Tam jest połączony z błonami komórkowymi poprzez tak zwane proteoglikany.
Ma jednak szczególne znaczenie dla komórek śródbłonka naczyń krwionośnych, ponieważ może bezpośrednio kontrolować hydrolizę trójglicerydów w chylomikronach i VLDL. W celu pomiaru aktywności lipoproteazy wstrzykuje się heparynę. Heparyna usuwa wiązanie lipaz lipoproteinowych z proteoglikanów, dzięki czemu po wstrzyknięciu heparyny występuje zwiększone stężenie wolnych lipaz lipoproteinowych, które można określić na podstawie ich aktywności. Badanie to może między innymi określić niedobór lipazy lipoproteinowej.
Choroby i zaburzenia
Brak lipazy lipoproteinowej często prowadzi do poważnych problemów zdrowotnych. Jeśli lipazy lipoproteinowej jest za mało lub jej aktywność jest niewystarczająca z powodu defektu genetycznego, trójglicerydy w chylomikronach i VLDL mogą być tylko słabo rozbite lub wcale.
Niedobór lipazy lipoproteinowej może być przede wszystkim genetyczny, jak również, na przykład, wtórny do chemioterapii. Pierwotny niedobór LPL występuje rzadko i jest spowodowany autosomalnym recesywnym defektem genetycznym. Powstaje tak zwana chylomikronemia, która charakteryzuje się mleczną, kremową surowicą i określana jest jako hiperlipidemia typu I. Triglicerydy w chylomikronach nie są już rozkładane. W rezultacie często pojawiają się ciężkie trzustki z nietolerancją mleka i bólami brzucha.
Ponadto stale rozwijają się pękające ksantomy i hepatomegalia. Jedyne opcje leczenia to dieta niskotłuszczowa i brak alkoholu. Ta choroba jest często spowodowana mutacjami w genie LPL na chromosomie 8 lub w genie APOC2. Wtórna postać hiperlipidemii typu I występuje zwykle podczas chemioterapii i ma charakter tylko przejściowy.

.jpg)



.jpg)





.jpg)
.jpg)

.jpg)