Plik Zespół Mowata-Wilsona jest rzadkim, genetycznym zaburzeniem rozwojowym o szerokim zakresie objawów. Oprócz anomalii twarzy, jelit i narządów płciowych jako część wady genetycznej występują wady serca i zaburzenia rozwoju mózgu. Nieuleczalną dotychczas chorobę można leczyć jedynie objawowo.
Co to jest zespół Mowata-Wilsona?

© Jezper - stock.adobe.com
Plik Zespół Mowata-Wilsona to dość młody obraz kliniczny. Zjawisko zróżnicowane klinicznie zostało po raz pierwszy opisane przez Mowata i Wilsona w 1998 roku. Oprócz zaburzeń rozwojowych obraz kliniczny charakteryzuje małogłowie i zespół objawów choroby Hirschsprunga. Uważa się, że przyczyną choroby jest wada genetyczna.
Ogólnie objawy są niezwykle zróżnicowane. Do tej pory rzadko badano tę rzadką chorobę. W rezultacie do tej pory dostępnych jest niewiele opcji terapeutycznych. Nie ma definitywnego rozpowszechnienia, ponieważ zaburzenie to rzadko lub wcale nie mogło być zdiagnozowane dobrze w XXI wieku. Obecnie jest około 200 udokumentowanych pacjentów z zespołem.
przyczyny
Mutacja genu powoduje zespół Mowata-Wilsona. Według ostatnich badań gen ZFHX1B jest genem powodującym chorobę. Uważa się, że przyczyną wady genetycznej jest region 2q22 chromosomu. Dotknięty gen ma rozmiar około 70 kb i składa się w sumie z dziesięciu eksonów 1214 aminokwasów. Gen ten koduje białko SIP1, które jest aktywne jako modulator transkrypcji i bierze udział w embriogenezie.
W związku z tym embriogeneza osób dotkniętych chorobą jest zaburzona. Chorobowe anomalie genu mogą odpowiadać całkowitej delecji, repozycjonowaniu lub anomalii sekwencyjnej. Wada genetyczna jest przekazywana w dziedziczeniu autosomalnym dominującym. Wadliwy allel na dwóch homologicznych chromosomach jest wystarczający, aby przekazać chorobę dziedziczną.
Objawy, dolegliwości i oznaki
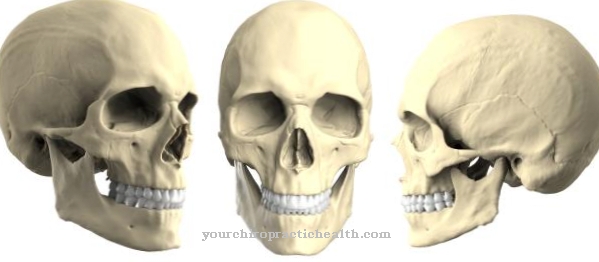
Objawy zespołu Mowata-Wilsona odpowiadają złożonemu zaburzeniu rozwojowemu i są zróżnicowane klinicznie. Główne objawy to drgawki wywołane mózgowo i małogłowie. Taka małogłowie występuje w wyniku przedwczesnego stwardnienia wszystkich szwów czaszkowych i obkurcza mózg w fazie wzrostu. Z tego powodu pacjenci doświadczają upośledzenia umysłowego. Ponadto często występują nieprawidłowości na twarzy, które często nadają pacjentowi profil podobny do orła.
Te anomalie mogą obejmować, na przykład, duże, głęboko osadzone oczy, poziomo ustawione brwi, nieprawidłowości w małżowinie usznej, wrastające płatki uszu i wyraźnie wystający podbródek. W 90 procentach przypadków osoby dotknięte chorobą cierpią na epilepsję. Rozwój umysłowy jest znacznie opóźniony, a rozwój językowy jest często całkowicie nieobecny. Spowolniony jest również rozwój motoryczny pacjenta.
Przy normalnych pomiarach urodzeń często występuje wtórny niski wzrost. Mogą wystąpić wady rozwojowe cewki moczowej. Możliwe są również wrodzone wady serca lub wady rozwojowe narządów płciowych. Ponadto występują nieprawidłowości neuronalne splotu ściany jelita, które są charakterystyczne dla choroby Hirschsprunga.
Diagnoza i przebieg choroby
Rozpoznanie zespołu Mowata-Wilsona nie może być postawione na podstawie samych badań, ale wymaga analizy materiału genetycznego. Laboratorium amplifikuje egzony od dwóch do dziesięciu genu ZFHX1B z genomowego DNA pacjenta. Ta amplifikacja odbywa się za pomocą PCR. Analiza tego materiału i miejsc splicingowych między intronem i egzonem odbywa się poprzez sekwencjonowanie DNA.
Każdy egzon genu ZFHX1B jest badany pod kątem delecji i duplikacji poprzez zależną od ligacji multipleksową amplifikację sondy. Ta skomplikowana procedura trwa około trzech tygodni i, w przeciwieństwie do samego badania pacjenta, może umożliwić postawienie jednoznacznej diagnozy. W większości przypadków, oprócz DNA jego rodziców, sekwencjonuje się i analizuje również DNA danej osoby.
Przebieg choroby zależy w dużej mierze od postaci nieprawidłowości genetycznej i zakresu delecji lub zmiany położenia części chromosomów. Trudno jest sformułować ostateczne prognozy ze względu na dotychczas mało udokumentowane przypadki zachorowań. Jednak wczesna diagnoza i późniejsza terapia prawdopodobnie wpłyną pozytywnie na rokowanie.
Komplikacje
Zespół Mowata-Wilsona powoduje u pacjenta poważne dolegliwości i powikłania, które znacznie skracają oczekiwaną długość życia i jego jakość. Z reguły codzienne życie pacjenta jest również znacznie ograniczone, a osoby dotknięte chorobą są w codziennym życiu zależne od pomocy innych osób.
Ponadto istnieje upośledzenie umysłowe, w którym krewni i rodzice często cierpią na dolegliwości psychiczne lub depresję. W większości przypadków osoby dotknięte chorobą cierpią również na skurcze i zmniejszoną odporność. Ponadto występują również różne deformacje twarzy i pojawia się epilepsja.
Rozwój językowy jest również znacznie opóźniony, przez co w wieku dorosłym występują znaczne trudności w porozumiewaniu się z pacjentem. Występuje również wada serca i niski wzrost. Wada serca może prowadzić do samoistnej śmierci sercowej, tak więc oczekiwana długość życia chorej osoby jest ograniczona przez zespół Mowata-Wilsona.
Nie ma lekarstwa na zespół Mowata-Wilsona. Jednak różne skargi można ograniczyć i rozpatrzyć, aby dana osoba miała znośne życie codzienne. Nie ma żadnych komplikacji, ale pozytywne leczenie nie zawsze jest możliwe.
Kiedy powinieneś iść do lekarza?
Chociaż zespołu Mowata-Wilsona nie można wyleczyć za pomocą obecnych możliwości prawnych i medycznych, leczenie występujących objawów może przynieść znaczną ulgę. Zwykle im wcześniej można postawić diagnozę, tym lepsze są możliwości terapeutyczne pacjenta. W przypadku wystąpienia zaburzeń rozwojowych u rosnącego dziecka konieczna jest konsultacja z lekarzem.
Jeśli istnieją indywidualne nieprawidłowości w bezpośrednim porównaniu z tymi w tym samym wieku, potrzebny jest lekarz. Obserwacje należy z nim omówić, aby możliwa była ocena stanu zdrowia. Lekarz powinien mieć problemy z uczeniem się, upośledzeniem pamięci, opóźnieniem mowy lub osobliwościami sekwencji ruchowych. W przypadku wystąpienia skurczów, bólu lub nieprawidłowej postawy, należy zgłosić się do lekarza. Wady rozwojowe lub nieprawidłowości twarzy wskazują na stan wymagający leczenia.
Wada wzroku lub nieprawidłowości w rysach twarzy powinien wyjaśnić lekarz. Spowolnione procesy myślowe lub ruchy są oznakami zaburzenia i należy je zbadać. W przypadku zaburzeń rytmu serca, problemów z wydalinami lub nieprawidłowości w zdolności reagowania lub postrzegania, należy skonsultować się z lekarzem. Zaburzenia behawioralne, zaburzenia wegetatywne lub osobliwości wyglądu skóry muszą zostać zbadane przez lekarza.
Leczenie i terapia
Zespół Mowata-Wilsona jest jak dotąd nieuleczalny. Opcje leczenia objawowego są również ograniczone. Terapie lecznicze są zwykle stosowane przeciwko napadom. Największą skuteczność w tym kontekście wykazują leki przeciwpadaczkowe. Niektóre objawowe wady rozwojowe można skorygować chirurgicznie. W szczególności objawy choroby Hirschsprunga należy korygować jak najwcześniej, ponieważ w przeciwnym razie może dojść do posocznicy lub zapalenia otrzewnej.
Leczenie objawowe zespołu Mowata-Wilsona ma przede wszystkim na celu poprawę jakości życia osób dotkniętych chorobą. W tym celu można również przeciwdziałać upośledzeniu umysłowemu i motorycznemu. Terapie logopedyczne mogą w pewnych okolicznościach pomóc w rozwoju języka, który w zespole Mowat-Wilson często kończy się niepowodzeniem bez wspomagających środków terapeutycznych. Zabiegi fizjoterapeutyczne i terapia zajęciowa mogą przeciwdziałać opóźnionemu rozwojowi motoryki.
Zespół Mowata-Wilsona jest często prawie niewyobrażalnym obciążeniem psychicznym dla rodziców osoby dotkniętej chorobą. Z tego powodu rodzice pacjentów są często wspierani przez psychoterapeutów. Obecnie badania medyczne dotyczą podejść do terapii genowej, która powinna leczyć defekty genów w przyszłości. W ten sposób wadliwy gen ZFHX1B u osób dotkniętych chorobą może wkrótce zostać zastąpiony, co może spowodować wyleczenie choroby.
Perspektywy i prognozy
Zespół Mowata-Wilsona można obecnie dobrze leczyć. Oczekiwana długość życia i jakość życia zależą od rodzaju i ciężkości wrodzonych wad rozwojowych. W przypadku łagodnych nieprawidłowości, które nie wpływają na serce, osoby dotknięte chorobą mogą dożyć dorosłości.
Poważnie chorzy pacjenci najczęściej umierają w wyniku choroby w dzieciństwie lub w okresie dojrzewania. Typowymi przyczynami zgonu są zawał mięśnia sercowego lub charakterystyczne choroby HSCR. Napady mózgowe często kończą się śmiercią w pierwszych latach życia dziecka. Rzadki zespół można leczyć objawowo, co oznacza, że pacjenci mogą przynajmniej czasowo prowadzić życie wolne od objawów.
Jednak w dłuższej perspektywie zespół Mowata-Wilsona nie daje pozytywnego rokowania, ponieważ różne wady rozwojowe i anomalie powodują postępujące pogorszenie stanu zdrowia i ostatecznie prowadzą do śmierci. Prognozy dotyczące oczekiwanej długości życia i przebiegu choroby zwykle sporządza odpowiedzialny specjalista. Najczęściej jest to neurolog lub specjalista chorób genetycznych. W zależności od objawów rozpoznanie choroby może być trudne, dlatego często zespół Mowata-Wilsona nie jest diagnozowany, zanim choroba nie będzie dobrze zaawansowana.
zapobieganie
Ponieważ zespół Mowata-Wilsona jest złożonym zaburzeniem rozwojowym o podłożu genetycznym, trudno jest temu zapobiec. Jednak pary zaangażowane w planowanie rodziny mogą poddawać sekwencjonowanie swojego DNA w celu oceny osobistego ryzyka przeniesienia wad genetycznych.
Opieka postpenitencjarna
W większości przypadków osoby dotknięte zespołem Mowata-Wilsona nie mają dostępnych środków kontrolnych lub mają ich tylko kilka, ponieważ jest to choroba genetyczna. Dlatego osoby dotknięte chorobą powinny najlepiej skonsultować się z lekarzem na wczesnym etapie, aby nie było dalszych dolegliwości lub komplikacji, które mogłyby mieć negatywny wpływ na długość życia i jakość życia danej osoby.
Z reguły nie dochodzi do samoleczenia, dlatego przy pierwszych oznakach i objawach choroby należy skonsultować się z lekarzem. Jeśli chcesz mieć dzieci, testy genetyczne i poradnictwo mogą być przydatne, aby zapobiec nawrotom zespołu u twoich potomków. Z reguły osoby dotknięte zespołem Mowata-Wilsona są uzależnione od przyjmowania różnych leków.
Należy je zawsze przyjmować w odpowiednim czasie i we właściwej dawce, aby złagodzić objawy. W przypadku dzieci szczególnie rodzice powinni kontrolować spożycie. W wielu przypadkach konieczne są również środki fizjoterapeutyczne, chociaż niektóre ćwiczenia można wykonywać również we własnym domu. Nie można jednoznacznie przewidzieć, czy zespół Mowata-Wilsona doprowadzi do skrócenia oczekiwanej długości życia osoby dotkniętej chorobą.
Możesz to zrobić sam
Ponieważ niestety nie ma lekarstwa na zespół Mowata-Wilsona, głównym priorytetem jest obecnie poprawa jakości życia dziecka.
W wielu przypadkach wcześnie rozpoczęta terapia logopedyczna może przeciwdziałać opóźnionemu rozwojowi językowemu i zapewnić znaczący sukces w rozwoju języka. Ponadto intensywne zabiegi fizjoterapeutyczne i terapia zajęciowa zapewniają lepszy rozwój motoryczny i umysłowy. Oprócz środków przepisanych przez lekarza wskazane jest również samodzielne zajęcie się tematem i kontynuowanie terapii w domu.
Opieka nad dzieckiem niepełnosprawnym jest ogromnym obciążeniem, zwłaszcza dla rodziców, ale także dla rodzeństwa, które może być obecne, co może mieć wpływ na życie rodzinne i ostatecznie na jakość opieki. Dlatego niezwykle ważne jest, aby w takich przypadkach rodzice w odpowiednim czasie szukali psychoterapii, która doda im sił na dłuższą metę poprzez naukę metod relaksacji i radzenia sobie z konfliktami.
Należy również pamiętać, że poszkodowani mają prawo do opieki profilaktycznej trwającej do sześciu tygodni każdego roku, za którą koszty pokrywa ubezpieczenie pielęgnacyjne. Istnieją już obiekty, które zapewniają intensywną opiekę w ciągu dnia, podczas gdy krewni mogą odpocząć podczas wycieczek. Może to być bardzo pomocne, zwłaszcza w przypadku rodzeństwa.











.jpg)