Plik Synteza hemoglobiny składa się z syntezy hemu i syntezy globiny. Wreszcie grupa prostetyczna hemu, każda z czterema globinami, jest połączona z kompleksem białkowym zawierającym żelazo - hemoglobiną. Zaburzenia w syntezie hemu i globiny mogą prowadzić do poważnych problemów zdrowotnych.
Co to jest synteza hemoglobiny?

Aby zrozumieć syntezę hemoglobiny, konieczna jest przede wszystkim znajomość budowy hemoglobiny. Hemoglobina to kompleks białkowy zawierający żelazo, który składa się z czterech podjednostek globiny, z których każda ma prostetyczną grupę hemu.
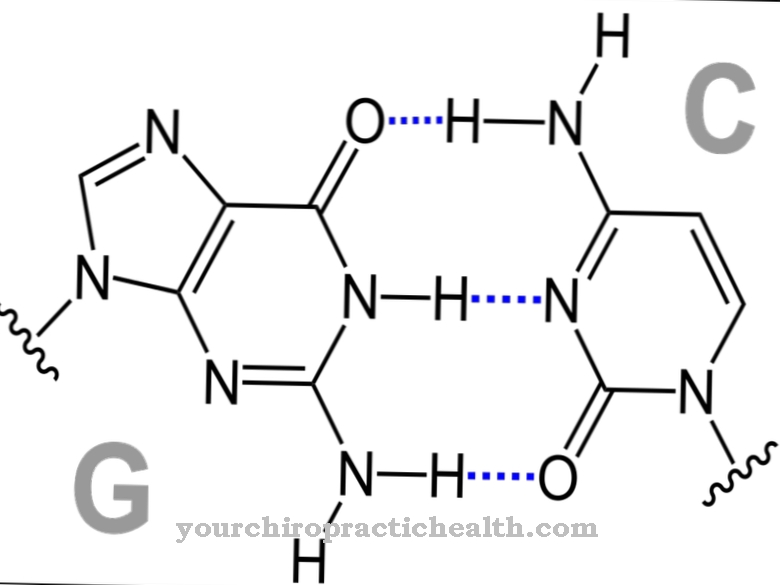
W ludzkiej hemoglobinie dorosłych występują dwie identyczne alfa-globiny, a także dwie identyczne beta-globiny jako podjednostki. Każda z tych podjednostek jest związana z protetyczną grupą hemu, która składa się z kompleksu porfiryny żelaza (II). Zatem kompleks hemoglobiny zawiera cztery grupy hemu.
W zależności od środowiska chemicznego każda grupa hemu może wiązać cząsteczkę tlenu z jonem żelazawym w złożony sposób. W zależności od tego, ile grup hemowych jest obciążonych tlenem, mówi się o oksyhemoglobinie (o wysokiej zawartości tlenu) lub deoksyhemoglobinie (o niskiej zawartości tlenu).
Jon żelazawy znajduje się w środku pierścienia porfirynowego. Z boku znajduje się złożone wiązanie z resztą histydynową globiny. Z drugiej strony, w zależności od stanu energetycznego jonu żelaza, cząsteczka tlenu może być związana w kompleksie. Na stan energetyczny wpływają zewnętrzne warunki fizyczne i chemiczne z powodu zmian konformacji globiny.
Funkcja i zadanie
Ostatni krok w syntezie hemoglobiny polega na złożeniu protezy grupy hemu z czterema jednostkami globiny w celu utworzenia kompleksu białkowego zawierającego żelazo. Poszczególne składniki są tworzone przez niezależne szlaki biosyntetyczne.
Materiałami wyjściowymi dla pierścienia porfirynowego grupy hemu są aminokwasy glicyna i sukcynylo-CoA. Sukcynylo-CoA składa się z koenzymu A i kwasu bursztynowego. Kwas bursztynowy jest produktem pośrednim w rozkładzie bogatych w energię ciał ketonowych w ramach metabolizmu energetycznego. Przy pomocy enzymu syntazy kwasu delta-aminolewulinowego, kwas delta-aminolewulinowy jest syntetyzowany z sukcynylo-CoA i glicyny. Dwie cząsteczki kwasu delta-aminolewulinowego kondensują się z eliminacją jednej cząsteczki wody, tworząc porfobilinogen, pochodną pirolu. Wraz z eliminacją amoniaku i przy pomocy enzymu syntetazy uroporfirynogenu-I, cztery cząsteczki porfobiliogenu reagują, tworząc hydroksymetylobilan. Jest on przekształcany w uroporfirynogen III z utworzeniem pierścienia.
Protoporfiryna jest wytwarzana w mitochondriach poprzez enzymatyczną dekarboksylację i odwodnienie. W przypadku enzymu ferrochelataza jon żelaza (II) jest włączany do tej cząsteczki z utworzeniem hemu. W cytozolu komórki hem jest połączony z białkiem globiną, tworząc kompleks białkowy zawierający żelazo - hemoglobinę.
Synteza poszczególnych globin odbywa się poprzez normalną biosyntezę białka. Jak już wspomniano, kompleks hemoglobiny dla dorosłych zawiera dwie identyczne podjednostki globin alfa i beta. Dzięki swojej złożonej budowie, gotowa hemoglobina rozwinęła zdolność transportu tlenu i dostarczania go do wszystkich komórek organizmu.
Jednak wiązanie centralnego żelaza z tlenem nie jest zbyt mocne i może bardzo łatwo wpływać na zewnętrzne czynniki chemiczne i fizyczne. Umożliwia to hemoglobinie szybkie wchłanianie i uwalnianie tlenu. Zawartość tlenu w hemoglobinie zależy między innymi od czynników pH, dwutlenku węgla lub ciśnienia parcjalnego tlenu lub temperatury. Te wpływające zmienne zmieniają, na przykład, zgodność globin, tak że wiązanie tlenowe może zostać wzmocnione lub osłabione przez niewielkie zmiany w warunkach energetycznych i sterycznych.
Przy niskiej wartości pH i wysokim ciśnieniu parcjalnym dwutlenku węgla wiązanie tlenu z jonem żelaza (II) jest osłabione, a tym samym sprzyja uwalnianiu tlenu. Dokładnie w tych warunkach następuje silniejszy obrót metaboliczny, który również powoduje zwiększone zapotrzebowanie na tlen. Dzięki funkcji hemoglobiny system transportu tlenu jest zatem optymalnie skoordynowany z potrzebami fizycznymi.
Choroby i dolegliwości
Zaburzenia w syntezie hemoglobiny mogą prowadzić do różnych chorób. Istnieje wiele chorób genetycznych, których podstawą jest zakłócenie syntezy hemu. W tym czasie w organizmie gromadzą się prekursory hemu, co między innymi prowadzi do ekstremalnej wrażliwości na światło. W tak zwanych porfiriach porfiryny są gromadzone w naczyniach krwionośnych, a nawet w wątrobie. Pod wpływem światła niektóre formy porfirii magazynują więcej energii promieniowania. Po uwolnieniu energii powstają rodniki tlenowe, które atakują i niszczą odsłoniętą tkankę. Prowadzi to do silnego swędzenia i piekącego bólu.
Istnieje siedem form porfiru. Budowa hemu to ośmiostopniowy proces, w którym zaangażowanych jest siedem enzymów. Jeśli enzym działa tylko nieprawidłowo, odpowiedni prekursor jest w tym momencie magazynowany w syntezie hemu. Na podstawie objawów porfirie dzieli się na dwie główne grupy. Tak zwane porfirie skórne charakteryzują się bolesną wrażliwością skóry na światło.W porfirii wątrobowej przeważa zajęcie wątroby z silnym bólem brzucha, nudnościami i wymiotami. Jednak w wielu przypadkach oba zespoły objawów pokrywają się.
Porfirie często mają przerywany przebieg z ostrymi atakami. W zależności od rodzaju porfirii objawiają się one nagle bolesnymi reakcjami skórnymi, kolkowym bólem brzucha, nudnościami / wymiotami, czerwonym zabarwieniem moczu, drgawkami, deficytami neurologicznymi, a nawet psychozami.
Inne zaburzenia syntezy hemoglobiny dotyczą nieprawidłowej syntezy cząsteczek globiny poprzez mutacje w odpowiednich genach. Przykładami są tak zwana anemia sierpowata lub talasemia. W niedokrwistości sierpowatokrwinkowej białko podjednostki beta globiny jest modyfikowane genetycznie. W szóstej pozycji tego białka aminokwas glutaminowy został zastąpiony waliną. Jeśli brakuje tlenu, hemoglobina przybiera kształt sierpa, zbija się razem i zatyka małe naczynia krwionośne. Skutkuje to zagrażającymi życiu zaburzeniami krążenia. Talasemie to grupa różnych wad rozwojowych hemoglobiny, które prowadzą do zmniejszonego tworzenia się łańcucha globiny alfa lub beta globiny. Najważniejszym objawem jest ciężka niedokrwistość.





.jpg)





.jpg)
.jpg)

.jpg)