Hipoksantyna jest odchyleniem purynowym i występuje w postaci związanej jako zasada nukleinowa oraz w postaci wolnej, np. B. w moczu. Występuje również w gruczołach i szpiku kostnym. Jako produkt deaminacji adeniny, hipoksantyna jest utleniana do kwasu moczowego i ksantyny. Rzadziej tworzy podstawową strukturę kwasów nukleinowych.
Co to jest fosforybozylotransferaza hipoksantyna guaniny?
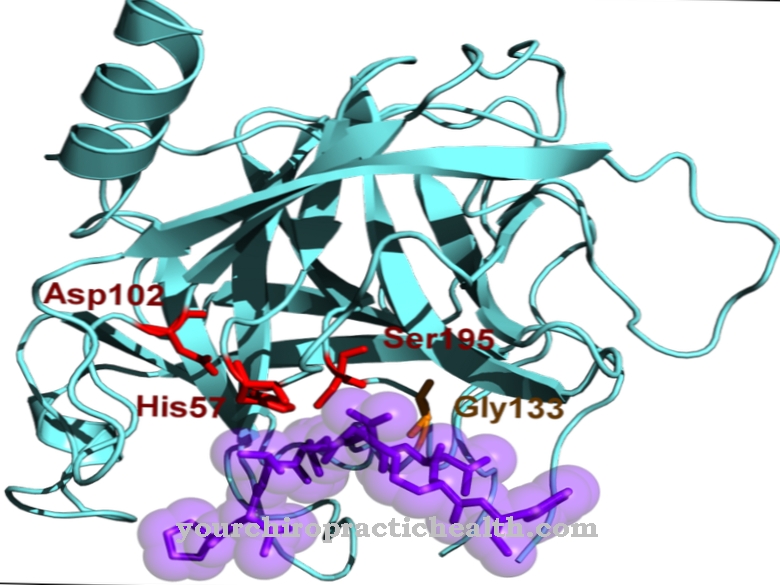
Enzym tetrameryczny powstaje z hipoksantyny i guaniny Hipoksantynowa fosforybozylotransferaza guaninowa.
Tetramery to makrocząsteczki, które składają się z czterech podobnych bloków budulcowych, a dokładniej z monomerów. Enzym ten jest jednym z najważniejszych w metabolizmie puryn u eukariotów, jest wrażliwy na zmiany genów i może powodować odchylenia u ludzi poprzez mutacje genów, które są wyrażane w niektórych chorobach metabolicznych. Takie są z. B. zespoły Lescha-Nyhana i Kelleya-Seegmillera.
Funkcja, efekt i zadania
Enzym hipoksantyno-guaninowo-fosforybozylotransferaza zwiększa metabolizm puryny i jej efektywność energetyczną.
Jest to oparte na zasadach purynowych, które są kwasami nukleinowymi, które strukturalnie pochodzą z puryny. Są to ksantyna, hipoksantyna, adenina i guanina, które łączą się z innymi zasadami poprzez wiązania wodorowe. Takie wiązania mają duży wpływ na podwójną helisę DNA i replikację oraz odgrywają rolę w biosyntezie białek.
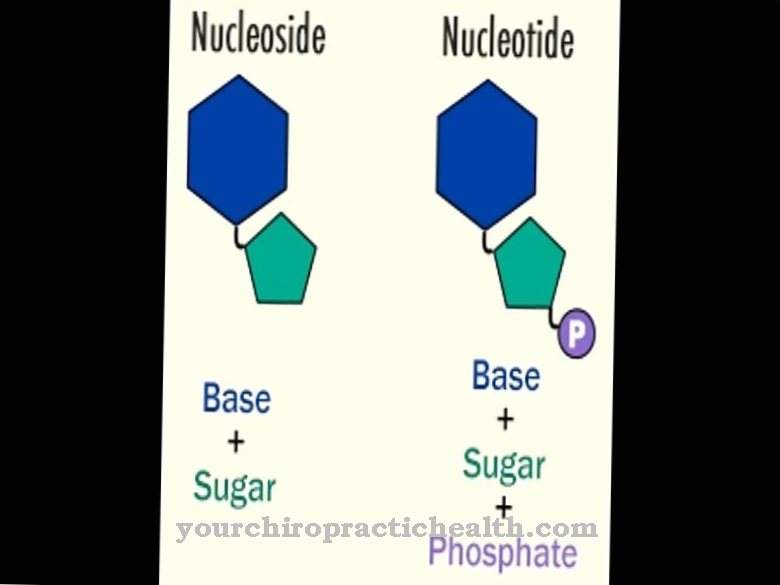
Zasady purynowe mogą być przetwarzane przez dwa enzymy. Oprócz hipoksantynowo-guaninowej fosforybozylotransferazy jest to fosforybozylotransferaza adeninowa. Obie tworzą nukleotyd poprzez resztę fosforybozylu, która z kolei jest podstawowym budulcem kwasów nukleinowych zarówno w DNA, jak i RNA. Cząsteczka składa się z cukru, zasady i fosforanu i kontroluje ważne funkcje regulacyjne w komórkach. Podczas gromadzenia się ATP jest oszczędzane, a tworzenie kwasu moczowego jest zmniejszone.
Jeśli zasady purynowe są poddawane recyklingowi, nazywa się to ścieżką odzysku. Jest to ogólny termin określający szlaki metaboliczne, w których synteza biomolekuł powstaje z produktów degradacji. Organizm przeprowadza własny proces recyklingu, w którym około dziewięćdziesiąt procent zasad purynowych jest ponownie wykorzystywanych, a dziesięć procent jest faktycznie wydalane. Pokazuje to skuteczność recyklingu zasady purynowej i znaczenie transferazy fosforybozylotransferazy hipoksantynowo-guaninowej.
Edukacja, występowanie, właściwości i optymalne wartości
Jeśli w genie HPRT wystąpią mutacje, rozmiar i aminokwasy mogą się zmienić. Może to być włączenie dodatkowych sekwencji DNA lub nukleotydów, co z kolei prowadzi do nieprawidłowego wytwarzania produktu genu, który jest kodowany na odpowiednim genie, lub nawet do delecji całej sekwencji. Czy z. B. zmienia sekwencję aminokwasów, pojawiają się choroby takie jak dna.
Szczególnie poważne są choroby metaboliczne, takie jak zespół Lescha-Nyhana w wyniku defektu genetycznego. Jest to dziedziczone w sposób recesywny sprzężony z chromosomem X, co oznacza, że dotyka głównie mężczyzn, którzy mają tylko jeden chromosom X. Wada genetyczna może występować u kobiet, ale ujawnia się jako choroba tylko wtedy, gdy dotyczy to obu chromosomów X, co jest stosunkowo rzadkie. Najczęściej drugi chromosom X kompensuje wadę pierwszego.
Tutaj znajdziesz swoje leki
➔ Leki na zdrowie pęcherza i dróg moczowychChoroby i zaburzenia
Zespół objawia się niedoborem hipoksantynowo-guaninowej fosforybozylotransferazy. Enzym nie jest produkowany z powodu wady genetycznej. Ze względu na mutację oraz brak recyklingu i konwersji zasad guaniny i hipoksantyny następuje nagromadzenie zasad purynowych, które muszą być gromadzone i wydalane przez organizm.
Rozkład zachodzi poprzez produkt pośredni ksantynę, która jest przekształcana w kwas moczowy i wydalana przez nerki. Jeśli ten proces jest ograniczony, w okolicy stawów tworzą się kryształy kwasu moczowego, które następnie wywołują więcej ataków dny moczanowej. Enzym nie jest już produkowany, wzrasta poziom kwasu moczowego w tkankach i krwi, a ośrodkowy układ nerwowy jest zaburzony.
Zespół Lescha-Nyhana nie jest bezpośrednio widoczny po urodzeniu. Wyraźne ułożenie nóg i tendencję dziecka do niewielkiego ruchu i wolniejszego rozwoju można zauważyć dopiero po około dziesięciu miesiącach. Zespół jest słaby i ciężki. Zwiększone wydzielanie kwasu moczowego i lżejsze ataki dny moczanowej są łagodniejszą postacią, z poważnymi objawami samookaleczenia, poważnymi zaburzeniami psychicznymi i agresją. Samookaleczenie następuje poprzez ugryzienie palców lub warg. Podczas gryzienia w kończyny często można zaobserwować, że osoby dotknięte chorobą ograniczają swoją autoagresję do jednej ręki. Z kolei agresja jest często skierowana przeciwko bliskim Ci osobom, takim jak rodzeństwo czy rodzice.
Najpoważniejsza postać choroby charakteryzuje się wieloma dysfunkcjami neurologicznymi i bardzo wyraźną skłonnością do samookaleczeń. Zespół objawia się spastycznością, dystonią, hipotonią, choreoatetozą i zwiększoną skłonnością do reagowania na odruchy. Cechy umysłowe i rozwój są poważnie ograniczone. W tym stanie zespół może również prowadzić do śmierci w szczególnie drastycznym stopniu.
Choroba jest diagnozowana na podstawie obrazu medycznego. Mierzy się poziom kwasu moczowego w moczu i krwi oraz aktywność fosforybozylotransferazy hipoksantynowo-guaninowej w tkankach i krwi. Ta ostatnia jest znacznie zmniejszona i może również występować w okresie prenatalnym.
Leczenie choroby jest trudne. Wyleczenie nie jest możliwe i bez leczenia dziecko umiera w pierwszych latach życia. W niektórych przypadkach zęby mleczne należy usunąć jako środek zapobiegawczy. Inne podejścia terapeutyczne obejmują obniżanie poziomu kwasu moczowego za pomocą leków, takich jak allopurynol, który działa jako inhibitor dny. Zasady purynowe nie są poddawane recyklingowi, ale kwas moczowy rozkłada się lepiej. Odpowiednie schorzenia, infekcje i uszkodzenia nerwów są również leczone i zalecana jest specjalna dieta, która zwykle nie zawiera mięsa i ma niską zawartość puryn.
Prowadzone są również badania nad psychosomatycznymi skutkami ubocznymi głębokiej stymulacji mózgu. Medycyna ma nadzieję, że zapobiegnie to agresji i samookaleczeniom. Z drugiej strony, zespół Kelleya-Seegmillera jest najłagodniejszą formą niedoboru fosforybozylotransferazy hipoksantynowo-guaninowej. Tutaj również wytwarza się zbyt dużo kwasu moczowego i pojawiają się wczesne choroby dny moczanowej. Pierwsze oznaki zespołu to pomarańczowe kryształy w pieluszce dziecka, infekcje dróg moczowych i kamica moczowa. Dna lub ostre zapalenie stawów rozwija się w okresie dojrzewania.
Niedorozwój umysłowy i autoataki, które występują przy zespole Lescha-Nyhana, nie mają miejsca, co najwyżej mogą prowadzić do zaburzeń uwagi. Wczesne leczenie zwykle umożliwia osobom dotkniętym chorobą normalną długość życia.




.jpg)





.jpg)
.jpg)

.jpg)