Oksydaza lizylowa jest enzymem tkanki łącznej, który ma zadania katalityczne i sprzyja sieciowaniu kolagenu i elastyny. Enzym działa stabilizująco na tkankę łączną, przeprowadzając deaminację oksydacyjną i tworząc w ten sposób podstawowe warunki do sieciowania. W Cutis laxa aktywność oksydazy lizylowej jest zmniejszona.
Co to jest oksydaza lizylowa?
W ludzkim ciele istnieją różne enzymy, z których wszystkie mają aktywność katalityczną. Enzymy umożliwiają lub przyspieszają reakcje w organizmie człowieka. Oksydaza lizylowa jest enzymem występującym w ludzkiej tkance łącznej. Jest również nazywany oksydazą białkową lizyny 6 i występuje głównie w przestrzeni zewnątrzkomórkowej tkanki łącznej.
Aktywność katalityczna enzymu w tym przypadku odnosi się do sieciowania między kolagenem i elastyną. Oksydaza lizylowa stabilizuje oba białka w sposób mechaniczny, umożliwiając w ten sposób reaktywne połączenie. Oksydaza lizylowa występuje nie tylko w organizmie człowieka. Inne kręgowce są również wyposażone w enzym. Uważa się, że oksydaza lizylowa jest stabilizatorem tkanki łącznej. Niedobór enzymu prowadzi do klinicznego obrazu cutis laxa, ciężkiego i dziedzicznego osłabienia tkanki łącznej.
Funkcja, efekt i zadania
Oksydaza lizylowa pełni ważne zadania w przestrzeni zewnątrzkomórkowej w połączeniu krzyżowym pomiędzy poszczególnymi cząsteczkami kolagenu. W ludzkim ciele kolagen odgrywa główną rolę w białkach, stanowiąc około 30 procent całkowitej masy białka.
Kolagen jest najpowszechniejszym białkiem. Jest białkiem strukturalnym i budulcowym, które tworzy wiele części ciała, takich jak tkanka łączna, kości, zęby, chrząstki, ścięgna, więzadła i skóra. Oksydaza lizylowa wspomaga wiązanie kolagenu z grupami karbonylowymi, a tym samym przyczynia się do stabilności wymienionych składników organizmu. Ma aktywność katalityczną do produkcji grup karbonylowych, które tworzą kowalencyjne wiązania krzyżowe na kolagenach w kondensacjach aldolowych. Zatem katalitycznym zadaniem oksydazy lizylowej jest przygotowanie do tworzenia włókienek. Enzym tworzy wszystkie warunki chemiczne niezbędne do powstania.
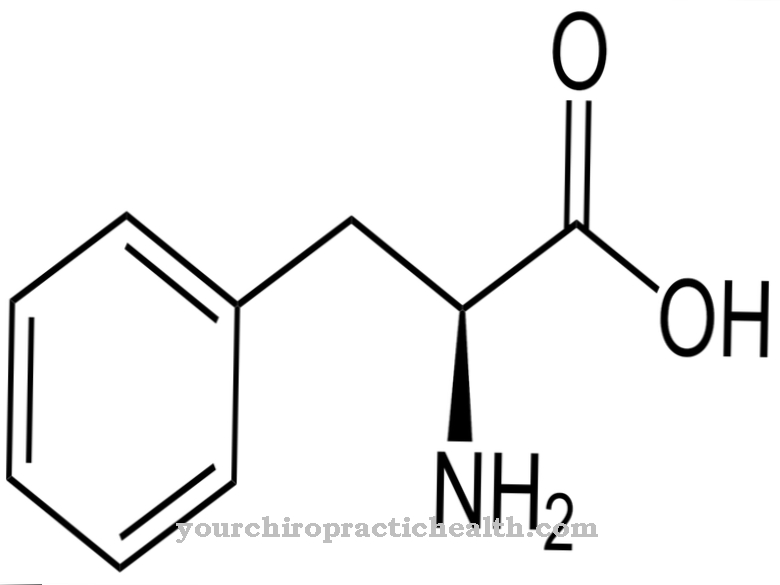
Fibryle są uważane za włókna błonnika. Odpowiadają one cienkim i włóknistym częściom ciała i znajdują się w ścianach komórek roślinnych, w mięśniach człowieka oraz w tkance łącznej. Zadaniem oksydazy lizylowej w tym kontekście jest zasadniczo oksydacyjna deaminacja reszt lizylowych. W chemii deaminacja to chemiczne odszczepienie grup aminowych w postaci jonów amonowych lub amoniaku. Dezaminacja oksydacyjna rozdziela grupy aminowe aminokwasu L-glutaminianu od wodoru i utlenia je do grup iminowych z przeniesieniem wodoru do NAD + lub NADP +.
Następnie następuje hydrolityczne rozszczepienie grup iminowych w postaci jonów amonowych, co jest związane z tworzeniem się α-ketokwasu. Deaminacja odpowiada pierwszemu etapowi biochemicznego rozkładu aminokwasów, który u ssaków zachodzi głównie w wątrobie. Jon amonowy powstały podczas deaminacji jest przekształcany w mocznik. W procesach deaminacji oksydazy lizylowej powstają grupy aldehydowe, które z poszczególnymi grupami aminowymi innych reszt lizylowych tworzą tak zwane zasady Schiffa i mogą w ten sposób tworzyć stabilizujące wiązania poprzeczne w kolagenie.
Edukacja, występowanie, właściwości i optymalne wartości
Oksydaza lizylowa w DNA jest kodowana przez gen LOX, który u ludzi jest zlokalizowany na chromosomie 5 w locus genu od q23.3 do q31.2. Produkt genu nie jest ostateczną formą enzymu. Produkt nie jest gotową oksydazą lizylową, ale jego poprzednią formą, która po translacji ma masę molową 47 kDa.
W dalszym przebiegu zachodzi glikozylacja. W trakcie tego procesu masa molowa późniejszego enzymu wzrasta do 50 kDa, a poprzednia forma oksydazy lizylowej jest wydzielana do przestrzeni zewnątrzkomórkowej. Po wydzieleniu oksydaza pre-pro-lizylowa jest dalej przetwarzana. Substancja jest rozszczepiana w przestrzeni zewnątrzkomórkowej. Za rozpad na dwa fragmenty odpowiada białko 1. W ten sposób z jednej strony powstaje oksydaza lizylowa 32 kDa. Z drugiej strony powstaje substancja resztkowa, która w tym przypadku odpowiada polipeptydowi.
Choroby i zaburzenia
Wady genetyczne oksydazy lizylowej mogą powodować obraz kliniczny cutix laxe. Ta choroba jest również nazywana dermatochalasis i odnosi się do grupy często związanych z wiekiem słabości tkanki łącznej, które w większości przypadków obserwuje się przy rodzinnej akumulacji.
Wspólną cechą wszystkich zjawisk dermatochalasis jest zwiotczenie i nieelastyczna skóra, która często zwisa w dużych fałdach na różnych częściach ciała. Większość osób dotkniętych chorobą wygląda na starszą niż w rzeczywistości z powodu zmian. Choroby te wywołują między innymi mutacje genetyczne. W tym kontekście mówimy o zespole cutis laxa. Choroba może występować w postaci autosomalnej recesywnej, autosomalnej dominującej i chromsomalnej x. W wielu przypadkach zespół cutis laxa jest powiązany z innymi anomaliami i, na przykład, jeśli zajęte są narządy, może być śmiertelny.
ARCL1 odpowiada cutis laxa typu 1 autosomalnego recesywnego i jest uważana za najcięższą postać, która może prowadzić do powikłań zagrażających życiu. Postać ARCL1A jest związana z mutacjami w genie FBLN5 w locus 14q32.12. Typ ARCL1B jest związany z mutacjami w genie EFEMP2 w locus 11q13.1, a wariant ARCL1C odpowiada cutis laxa z towarzyszącymi anomaliami w płucach, układzie pokarmowym i drogach moczowych, które są spowodowane mutacjami w genie LTBP4 w locus 19q13.2.
Mutacje w wymienionych genach prowadzą do poniżej średniej aktywności liksyloksydazy. Nieodpowiednie połączenia krzyżowe powstają z powodu zmniejszonej aktywności enzymu. Tkanka łączna pacjenta nie jest dostatecznie ustabilizowana.


.jpg)




.jpg)





.jpg)
.jpg)

.jpg)