Plik Syntaza ATP katalizuje syntezę ATP. Jest to kompleks składników hydrofobowych i hydrofilowych.
Co to jest syntaza ATP?

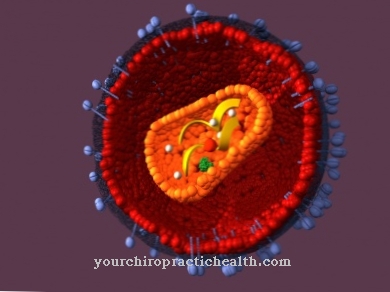
Syntaza ATP jest białkiem transbłonowym, które działa zarówno jako pompa protonowa, jak i enzym do syntezy ATP. Jako pompa protonowa wykorzystuje ATP do produkcji energii, podczas gdy wykorzystuje energię do budowy ATP.
Enzym składa się z 8 do 20 podjednostek, które grupują się razem, tworząc dwa kompleksy. Jeden kompleks jest nierozpuszczalny w wodzie i znajduje się w membranie. Ten kompleks transportuje protony na wyższe poziomy energii podczas zużywania ATP. Rozpuszczalna w wodzie część enzymu na wewnętrznej krawędzi błony katalizuje syntezę ATP z ADP przy użyciu gradientu protonów. Część nierozpuszczalną w wodzie określa się jako złożoną Fo, a część rozpuszczalną w wodzie jako kompleks F1.
Ze względu na dwie podjednostki nazwa ta jest również używana dla syntazy ATP FoF1-ATPase Podanie. Żaden inny enzym syntetyzujący ATP nie został jeszcze rozpoznany. Jest do tego wiele tak zwanych ATPaz, ale wszystkie używają tylko ATP. Syntaza ATP jest jedynym enzymem, który może zarówno konsumować, jak i syntetyzować ATP.
Funkcja i zadanie
Syntaza ATP odgrywa kluczową rolę w metabolizmie energetycznym. Wszystkie energochłonne procesy metaboliczne zależą od funkcji magazynowania energii ATP. Ze względu na dużą liczbę procesów metabolicznych każdego dnia w ludzkim organizmie syntetyzowane jest 80 kilogramów ATP i natychmiast ponownie rozkładane.
ATP jest budowany z ADP poprzez akumulację pozostałości fosforanu podczas pochłaniania energii. Tworzy to cząsteczkę naładowaną napięciem, która chciałaby szybko uwolnić swoją energię, uwalniając resztę fosforanu. Gdyby jednak nie było pośredniego magazynowania energii przez ATP, nie miałyby miejsca żadne procesy metaboliczne.
ATP występuje powszechnie w żywych organizmach. Dotyczy to roślin, grzybów i bakterii, a także ludzi i zwierząt. Jednak we wszystkich organizmach syntaza ATP zapewnia również tworzenie ATP. Przez długi czas mechanizm przekazywania energii nie był jasny. Większość naukowców zakładała, że energia pochodzi z wysokoenergetycznych związków pośrednich i jest przenoszona do ATP. Jednak brytyjski chemik Peter D. Mitchell jako pierwszy postulował, że ATP czerpie energię z gradientu protonów (gradient PH), przy czym główną rolę odgrywa syntaza ATP. Ta teza została później potwierdzona.
Zgodnie z ustaleniami naukowców synteza ATP zachodzi w rozpuszczalnej w wodzie części enzymu. Jeśli proton zostanie odszczepiony w nierozpuszczalnej w wodzie części enzymu w membranie, ujemny ładunek powstały podczas odszczepienia może zostać ustabilizowany poprzez oddziaływanie jonowe. Aby to zrobić, cząsteczka musi się trochę skręcić i wytworzyć napięcie. Syntaza ATP zgromadziła teraz energię. Kiedy ładunek ujemny jest protonowany, cząsteczka odskakuje jak napięta sprężyna i przenosi ruch obrotowy na zewnętrzną rozpuszczalną w wodzie część enzymu. Tam transfer energii odbywa się poprzez wychwyt reszty fosforanowej z ADP, dzięki czemu powstaje ATP. ATP to z kolei cząsteczka bogata w energię o wysokim napięciu mechanicznym, która uwalnia swoją energię, uwalniając resztę fosforanu.
Syntaza ATP jest nieodłączną częścią łańcucha oddechowego i występuje w dużej mierze w mitochondriach. Dzięki syntazie ATP odkryto jedyny enzym budujący ATP. Bez ATP wszystkie procesy energetyczne w organizmie zatrzymałyby się. Wraz z odkryciem syntazy ATP ostatecznie wyjaśniono niezrozumiany związek między utlenianiem NADH a syntezą ATP.
Tutaj znajdziesz swoje leki
➔ Leki na osłabienie mięśniChoroby i dolegliwości
W związku z syntazą ATP mogą również wystąpić choroby, które objawiają się głównie zaburzeniami w łańcuchu oddechowym. Produkcja energii w organizmie odbywa się w mitochondriach. Tam związki o wysokiej zawartości energii, takie jak kwasy tłuszczowe lub glukoza, są stale rozkładane w cyklu kwasu cytrynowego. Jako egzystencjalnie ważny krótkoterminowy magazyn energii, ATP musi być stale formowany.
Jeśli synteza ATP jest zakłócona, nie można uwolnić wystarczającej ilości energii. Pojawiają się objawy, takie jak masywne osłabienie i zmęczenie. Zawsze dotyczy to mięśni i układu nerwowego. Ponieważ procesy energetyczne zachodzą w mitochondriach, choroby te nazywane są również mitochondriopatiami.
Charakterystyczne dla tych zaburzeń są nieprawidłowe działanie lub uszkodzenie mitochondriów. Jednak przyczyny chorób mitochondrialnych są zróżnicowane. Istnieją dziedziczne i nabyte formy choroby. W dziedzicznych mitochondriopatiach mutacje enzymów występują w łańcuchu oddechowym. W przypadku choroby dziedzicznej zarówno mitochondrialne DNA, jak i DNA z jądra komórkowego mogą zostać zmienione przez mutacje. Oczywiście pośród wielu enzymów łańcucha oddechowego syntaza ATP może również podlegać mutacji.
Gdyby enzym całkowicie zawiódł, organizm nie byłby zdolny do życia. Dzięki ograniczonej funkcji można go zaliczyć do dużej grupy chorób mitochondrialnych. Różne formy chorób mitochondrialnych prowadzą do różnych wzorców objawów. Jednak wszyscy mają wspólne zaburzenia nerwowo-mięśniowe. Oznacza to, że niski poziom energii ma zawsze wpływ na układ nerwowy i mięśnie. Często dochodzi do upośledzenia układu sercowo-naczyniowego i nerek.
Ponieważ zaopatrzenie w energię jest zakłócone, większość chorób postępuje szybko. Jednak nie ma terapii przyczynowych, ponieważ jest to choroba genetyczna. Ważne jest zwiększenie spożycia energii w postaci węglowodanów i tłuszczów oraz ograniczenie zużycia energii.





-durch-vitamin-b12-mangel.jpg)