W którym Zespół Ehlersa-Danlosa, znane również jako EDS, to zaburzenia tkanki łącznej dziedziczone jako część wady genetycznej. Przede wszystkim EDS przejawia się poprzez nadmiernie ruchliwe stawy oraz nadmierną rozciągliwość skóry. Czasami EDS może również wpływać na naczynia, więzadła, mięśnie, ścięgna i narządy wewnętrzne; rokowanie zależy od rodzaju rozpoznanego zespołu.
Co to jest zespół Ehlersa-Danlosa?

© Astrid Gast - stock.adobe.com
Zespół Ehlersa-Danlosa jest bardzo rzadką chorobą tkanki łącznej. Jest to choroba dziedziczna, która występuje jako część wady genetycznej. Istnieje zaburzenie syntezy kolagenu. Ponieważ tkanka łączna jest obecna w całym ciele, objawy i oznaki zespołu Ehlersa-Danlosa mogą być bardzo różne i zróżnicowane.
Czasami osoba dotknięta chorobą może skarżyć się na nadmierną rozciągliwość skóry; w niektórych przypadkach mówi się nawet o pęknięciu naczyń i narządów wewnętrznych. Istnieje dziesięć typów EDS, które mają różne objawy i przebieg.
przyczyny
Zespół Ehlersa-Danlosa jest wywoływany przez defekt genetyczny w syntezie kolagenu. Około 80 procent wszystkich chorych cierpi na typy od I do III, typy od IV do VII stanowią tylko ułamek wszystkich chorych; Typy od VIII do X są jeszcze rzadsze. Podczas gdy typy I i II zespołu Ehlersa-Danlosa wpływają na kolagen V, typ III jest zaburzeniem związanym z kolagenem III.
Następujące geny mogą wywołać zespół Ehlersa-Danlosa: COL1A1 do COL3A1 oraz COL5A1 i COL5A2 i TNXB. Czasami za zespół mogą być również odpowiedzialne geny ADAMTS2 i PLOD1. W zależności od typu zależy również sposób dziedziczenia. Wiele postaci zespołu Ehlersa-Danlosa jest dziedziczonych jako cecha autosomalna dominująca.
Oznacza to, że tylko jeden z alleli musi zostać dotknięty zmianą, aby zespół Ehlersa-Danlosa wybuchł. Ale są też formy, które są dziedziczone jako cecha autosomalna recesywna. Oba allele muszą wykazywać zmianę, aby choroba mogła zostać przeniesiona.
Objawy, dolegliwości i oznaki
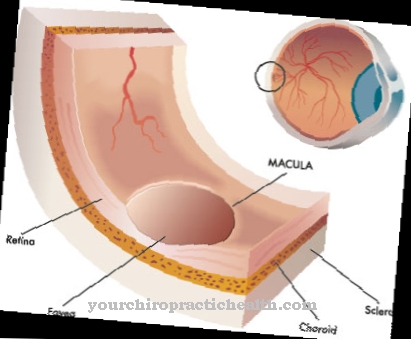
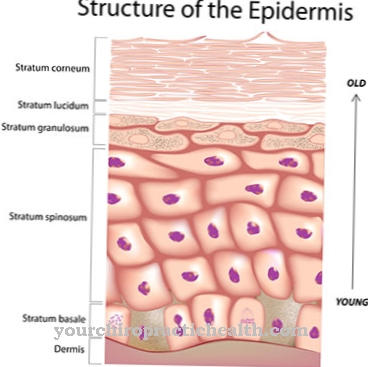
Ze względu na zaburzenia syntezy kolagenu niedostatecznie rozwinięte są naczynia krwionośne, skóra i stawy. Oznacza to, że pacjent cierpi na brak jędrności, aw konsekwencji na nadmierną rozciągliwość tkanki łącznej. Prowadzi to do lekkiego rozerwania już dotkniętych struktur. Przede wszystkim dochodzi do pękania naczyń krwionośnych, skrzywienia kręgosłupa czy pęknięć jelit. Czasami możliwa jest nawet nawracająca odma opłucnowa.
Przy typach I i II pacjent skarży się na bardzo łatwo zranioną i nadmiernie rozciągliwą skórę. Proces gojenia się ran jest bardzo nieprawidłowy, a stawy są bardzo mocne, aw niektórych przypadkach mogą również wpływać na naczynia krwionośne i narządy wewnętrzne.
W typie III choroba dotyczy tylko częściowo skóry; Przede wszystkim osoby dotknięte chorobą skarżą się na wyjątkowo wyraźną nadmierną ruchliwość stawów. Typ IV ma bardzo cienką, prawie przezroczystą skórę. Pacjent ma wyraźną skłonność do krwiaków, a także ma wyjątkowo przesuszone stawy, przez co na tym etapie często występuje zespół Ehlersa-Danlosa także na naczynia i narządy wewnętrzne.
Typ VI przede wszystkim skarży się na umiarkowaną nadmierną rozciągliwość skóry. Gojenie się rany jest nieprawidłowe; czasami zaangażowane są również narządy wewnętrzne. W wielu przypadkach dochodzi również do zajęcia oczu. W typie VII A i B skóra jest nadmiernie rozciągliwa, co czasami jest niezwykle cienkie. Wielu pacjentów skarży się również na zwichnięcie biodra. Z kolei w przypadku typu VII C pacjent zgłasza zwiotczenie skóry i zajęcie narządów wewnętrznych. Stawy są również hipermalne.
Diagnoza i przebieg
Lekarz stawia diagnozę kliniczną, odnosząc się przede wszystkim do wywiadu rodzinnego. Lekarz sprawdza również zwiększoną kruchość naczyń włosowatych za pomocą testu Rumpel-Leede, a także wykonuje biopsję skóry, po której następuje badanie mikroskopowe elektronowe całej struktury kolagenu. Typy można zidentyfikować za pomocą amplifikacji ludzkiego DNA genetycznego.
W zależności od rodzaju, rokowanie i przebieg choroby zespołu Ehlersa-Danlosa są różne. Podczas gdy niektórzy pacjenci mają bardzo niewiele ograniczeń, inni mają problemy w życiu codziennym. Przede wszystkim ból, ekstremalne niestabilności stawów i deformacje kręgosłupa powodują, że istnieje ogromne ograniczenie ruchomości.
Jednak większość pacjentów ma normalną długość życia; Tylko u nielicznych pacjentów, którzy cierpią również na upośledzenie naczyń krwionośnych, pojawiają się poważne komplikacje w kontekście zespołu Ehlersa-Danlosa.
Komplikacje
Powikłania w zespole Ehlersa-Danlosa są w dużym stopniu zależne od rozpoznanego typu. W ciężkich przypadkach dochodzi do deformacji skóry, mięśni i więzadeł. Może to również dotyczyć narządów wewnętrznych. Skóra pacjentów jest często nadmiernie rozciągnięta, a stawy mogą się nadmiernie poruszać.
W szczególności dzieci bardzo cierpią na zespół Ehlersa-Danlosa z powodu zastraszania i dokuczania. Może to prowadzić do depresji i problemów psychologicznych.
Skóra może ulec uszkodzeniu nawet przy niewielkich wpływach mechanicznych i jest łatwa do rozdarcia. Zwiększa to ryzyko wypadków i stanów zapalnych.
Na kręgosłup wpływa również zespół Ehlersa-Danlosa, który może prowadzić do skrzywienia kręgosłupa. Przy wszystkich typach czasami dochodzi do nieprawidłowego gojenia się ran, dlatego infekcje i stany zapalne należy leczyć ze szczególną ostrożnością. Leczenie lub terapia nie są możliwe w przypadku zespołu Ehlersa-Danlosa.
Jednak pacjent musi uważać, aby nie wykonywać żadnych czynności obciążających fizycznie. Podobnie rany i przeziębienia należy leczyć bardzo ostrożnie.Zespół Ehlersa-Danlosa nie prowadzi do skrócenia oczekiwanej długości życia. Powikłania mogą wynikać przede wszystkim ze skrzywienia kręgosłupa lub opóźnionego gojenia się ran.
Kiedy należy iść do lekarza?
Należy skonsultować się z lekarzem w przypadku częstych łez skóry, nieprawidłowego gojenia się ran i innych objawów wskazujących na upośledzenie tkanki łącznej. Zespół Ehlersa-Danlosa objawia się także krwiakami, nadmiernie wrażliwą skórą, a czasem także skrzywieniami kręgosłupa i pęknięciami jelit. W dalszym przebiegu mogą również wystąpić problemy z biodrami i choroby narządów wewnętrznych. Wszystkie te dolegliwości wymagają natychmiastowego wyjaśnienia lekarskiego.
Ponieważ zespół Ehlersa-Danlosa jest chorobą dziedziczną, warto przyjrzeć się historii rodziny. Przyszli rodzice, którzy sami cierpią na tę chorobę lub mają przypadki tej choroby w swoich rodzinach, powinni poddać dziecko badaniu natychmiast po urodzeniu.
Najpóźniej w przypadku wystąpienia poważnych powikłań, takich jak krwawienie lub silny ból, lekarz musi wyjaśnić przyczynę. Innymi kontaktami są dermatolog, różni interniści lub specjalista chorób dziedzicznych. W przypadku nagłego wypadku medycznego najlepiej jest natychmiast powiadomić pogotowie ratunkowe lub odwieźć poszkodowanego do najbliższego szpitala.
Lekarze i terapeuci w Twojej okolicy
Leczenie i terapia
Nie ma leczenia objawowego ani przyczynowego. Z tego powodu wszystko kręci się wokół profilaktyki wszelkich następczych szkód. Pacjent musi uważać, aby uniknąć urazów i dużego obciążenia fizycznego. Oznacza to, że należy unikać wszelkich sportów, które czasami zwiększają ryzyko kontuzji.
Ryzyko wzrasta w czasie ciąży; dlatego szczególnie osoby dotknięte typami I, II, IV i VI muszą podlegać ścisłemu monitorowaniu. Ważne jest również, aby mieć lek na kaszel w przypadku przeziębienia. Dzieje się tak, ponieważ można zapobiec pęknięciu jelita grubego, a następnie odmie opłucnowej. Jeśli pojawią się rany, należy je ostrożnie leczyć, aby przyspieszyć gojenie się ran.
Perspektywy i prognozy
Rokowanie w przypadku zespołu Ehlersa-Danlosa (EDS) zależy od rodzaju choroby. Jest również inny dla każdej osoby, której dotyczy. Pacjenci z hipermobilną postacią EDS mają zwykle normalną długość życia. W przypadku naczyniowego typu EDS istnieje większe ryzyko powikłań spowodowanych nagłym krwawieniem wewnętrznym. Jednak w obu formach jakość życia jest poważnie obniżona.
Ponadto obie formy zespołu Ehlersa-Danlosa obejmują kilka dziedzicznych chorób tkanki łącznej. Dlatego obecnie nie jest możliwa żadna terapia przyczynowa. Nawet możliwości leczenia objawowego są ograniczone. Dlatego ważne jest, aby pacjenci uważali, aby nie odnieść żadnych obrażeń. Nie należy wykonywać nawet przyziemnych operacji, aby uniknąć obrażeń. Są jednak pacjenci, którzy prawie nie cierpią z powodu ograniczeń. Inni są poważnie upośledzeni w życiu codziennym.
Ogólnie zespół Ehlersa-Danlosa ma charakter postępujący. W trakcie życia ograniczenia rosną. Mogą wystąpić niestabilności stawów, deformacje kręgosłupa i silny ból, co znacznie ogranicza mobilność. W naczyniowej postaci choroby istnieje również stałe ryzyko pęknięcia naczyń krwionośnych, co prowadzi do powikłań zagrażających życiu. Najgorszą konsekwencją EDS jest jednak poważne pogorszenie jakości życia spowodowane ograniczoną mobilnością i ciągłym lękiem przed bolesnymi pęknięciami.
zapobieganie
Nie ma żadnej profilaktyki. Czasami tylko badania w ramach diagnozy przedimplantacyjnej lub diagnozy ciała polarnego mogą zapobiec dziedziczeniu zespołu Ehlersa-Danlosa.
Opieka postpenitencjarna
W przypadku zespołu Ehlersa-Danlosa osoba dotknięta chorobą ma zwykle bardzo niewiele dostępnych środków kontrolnych lub nawet ich nie ma. Ponieważ jest to choroba wrodzona, nie można jej leczyć przyczynowo, a jedynie objawowo, aby nie nastąpiło całkowite wyleczenie. Z tego powodu wczesne wykrycie jest na pierwszym planie w zespole Ehlersa-Danlosa, dzięki czemu nie ma dalszych komplikacji i dolegliwości dla osoby zainteresowanej.
Im wcześniej zespół zostanie rozpoznany przez lekarza, tym zwykle jest lepszy, tym dalej przebiega. Jeśli pacjent chce mieć dzieci, można również przeprowadzić poradnictwo dziedziczne, aby zapobiec przenoszeniu zespołu na potomków. W większości przypadków zespół ten nie wpływa negatywnie na oczekiwaną długość życia pacjenta.
Jednak osoby dotknięte chorobą są bardzo zależne od pomocy przyjaciół i rodziny w życiu codziennym, aby móc radzić sobie w życiu codziennym. Naczynia krwionośne należy również regularnie sprawdzać, aby nie było pęknięć, które mogą zagrażać życiu osoby zainteresowanej. U dzieci z zespołem Ehlersa-Danlosa należy zapewnić wszechstronną edukację, aby uniknąć nieporozumień.
Możesz to zrobić sam
Pacjent nie może wyleczyć zespołu Ehlersa-Danlosa własnymi środkami pomocniczymi. Choroba dziedziczna jest uważana za nieuleczalną. Jednak w życiu codziennym można podjąć różne środki w celu złagodzenia objawów. Opcji wspomagających należy używać wielokrotnie, ponieważ nie można przy ich pomocy osiągnąć trwałej minimalizacji objawów.
Słabość tkanki łącznej może być zamaskowana modnymi cięciami odzieży i dodatków. Noszenie bielizny modelującej sylwetkę i stabilizującej, a jednocześnie nieco luźnej odzieży pomaga, aby inni ludzie nie widzieli zaburzeń tkanki łącznej w życiu codziennym. Masaże, kremy lub naprzemienne kąpiele pobudzają ukrwienie skóry i wspomagają organizm. Zajęcia sportowe pomagają budować mięśnie. W ten sposób można zminimalizować niektóre obszary słabości tkanki łącznej.
Rodzice i lekarze powinni szczegółowo wyjaśnić na temat zespołu Ehlersa-Danlosa i opisać jego objawy oraz przebieg choroby. Pełne poinformowanie dziecka przynosi korzyści. Cyfrowa wymiana z innymi chorymi może być również pomocna w radzeniu sobie z chorobą w życiu codziennym. Zaufanie do własnych kompetencji powinno być szczególnie wspierane i zachęcane na wczesnym etapie życia dziecka. Posiadanie silnej samooceny pomaga dziecku w zapewnieniu dobrej jakości życia z zespołem.

.jpg)




.jpg)





.jpg)
.jpg)

.jpg)